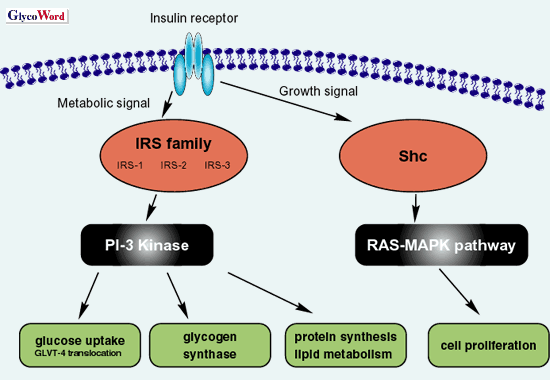
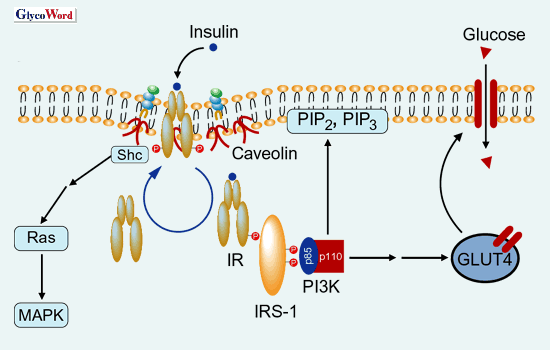
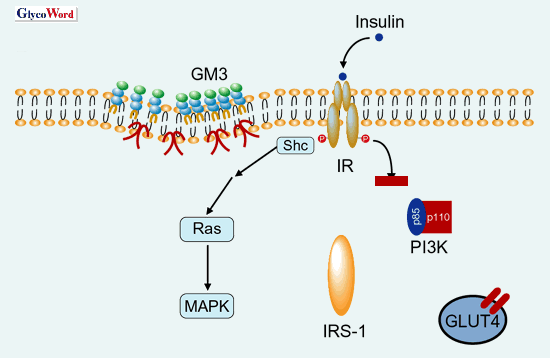
|
| References |
(1) |
Kadowaki, T. J. Clin. Invest. 106, 459-465 (2000) |
|
(2) |
Bremer, E. G., Schlessinger, J., and Hakomori, S. J.Biol.Chem.
261, 2434-2440 (1986) |
|
(3) |
Nojiri, H., Stroud, M., and Hakomori, S. J. Biol. Chem.
266, 4531-4537 (1991) |
|
(4) |
Gustavsson, J., Parpal, S., Karlsson, M., Ramsing, C., Thorn,
H., Borg, M., Lindroth, M., Peterson, KJ., Magnusson, K, and Stralfors,
P. FASEB J. 13, 1961-1971 (1999) |
|
(5) |
Tagami, S., Inokuchi, J., Kabayama, K., Yoshimura, H., Kitamura,
F,. Uemura, S., Ogawa, C., Ishii, A., Saito, M., Ohtsuka, Y., Sakaue,
S., and Igarashi, Y.
J. Biol. Chem. 277, 3085-2216 (2002) |
|
(6) |
Yamashita, T., Hashiramoto, A., Haluzik, M., Mizukami, H., Beck,
S., Norton, A., Kono, M., Tsuji, S., Daniotti, J.L., Werth, N.,
Sandhoff, R., Sandhoff, K., and Proia, R.L. Proc. Natl. Acad,
Sci. USA, 100, 3445-3449(2003) |
|
|
|
|
|