

|
|
Genetic Disease Caused by Incomplete Processing of N-glycans | |||||||||||||||||
 |
Introduction Congenital dyserythropoietic anemia type II, or HEMPAS (hereditary erythroblastic multinuclearity with positive acidified serum lysis test), is a genetic anemia in humans. Erythrocyte membrane glycoproteins, such as band 3 and band 4.5, which are normally glycosylated with polylactosamines lack polylactosamines, in HEMPAS. Instead, polylactosamines accumulate as glycolipids in HEMPAS. Analysis of N-glycans from HEMPAS erythrocyte membranes revealed a series of incompletely processed N-glycan structures, and characterize this disease as N-glycan processing deficiency (1). Incidence HEMPAS is an autosomal recessive anemia. Parents and some siblings of HEMPAS patients are clinically normal but are heterozygous carriers of the defective gene. The numbers of patients reported in the literature is 130, although as many as 300 patients may exist. HEMPAS patients have been found on all continents and in a variety of races. Both sexes are equally affected. Various case reports on HEMPAS patients suggest that the disease is often asymptomatic and therefore not as rare as hitherto believed. This disease may become clinically apparent when another hereditary or acquired disease occurs. Symptoms HEMPAS patients suffer from life-long anemia. Jaundice is common. In most cases the degree of anemia is mild; however, there are severely affected patients who require constant care and frequent transfusions. The most striking diagnostic feature of HEMPAS is abnormal multinucleated erythroblasts in the bone marrow (Fig. 1). Erythrocytes released to the peripheral blood are short-lived: 7-34 days in HEMPAS versus 100 days in normal adults. The spleen may be the principal site of erythrocyte destruction, leading to splenomegaly in HEMPAS. Surgical removal of the spleen often alleviates the anemia. Also common are liver hemosiderosis and cirrhosis. Some HEMPAS patients need to withdraw blood in order to prevent hemosiderosis and cirrhosis. Severe cases of HEMPAS show mental and sensory abnormalities. | ||||||||||||||||
| Fig 1. Bone marrow aspirate from a HEMPAS patient showing multinucleated erythroblasts. 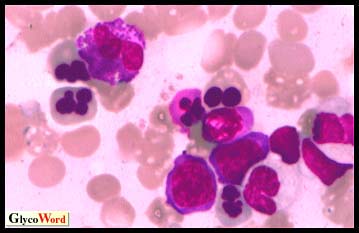
| |||||||||||||||||
|
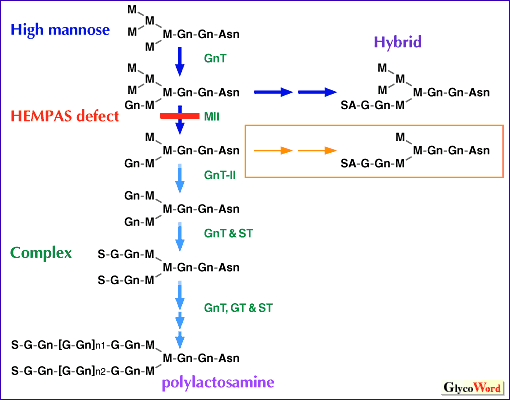
N-glycan processing defect in HEMPAS In normal erythrocytes, glycoproteins, such as band 3, are glycosylated by polylactosamine, a large carbohydrate chain made of galactose and N-acetylglucosamine repeats. In normal erythrocytes, the majority of glycolipids belong to the globo-series with short carbohydrate chains. In HEMPAS, glycoproteins lack polylactosamines whereas polylactosamines accumulate as glycolipids. Thus it appears that a genetic factor in HEMPAS blocks the glycosylation of glycoprotein acceptors and shifts polylactosamines to lipid acceptors. Structural analysis of HEMPAS erythrocyte N-glycans revealed the accumulation of hybrid type oligosaccharides and disruption of N-glycan synthesis around N-acetylglucosaminyltransferase II (GnT-II) and alpha-mannosidase II (MII) steps (Fig. 2). Analysis of N-glycan structures of HEMPAS serum glycoproteins revealed that these glycoproteins are incompletely processed. Thus it is clear that the glycosylation defect is not restricted to the erythroid lineage. Incompletely processed HEMPAS serum glycoproteins are recognized by hepatic lectin and cleared from the circulation. The quantity of incompletely processed serum glycoproteins which should be cleared by the liver is overwhelming, and this could be one of the reasons for liver cirrhosis in HEMPAS. | |||||||||||||||||
| |||||||||||||||||
|
Primary defect of HEMPAS HEMPAS is genetically heterogeneous. The first HEMPAS case identified as being defective in the MII gene is a patient from England. The patient cells express MII mRNA at less than 10% of normal levels (2). Recently another HEMPAS case (Irish) having mutations in the coding region of the MII gene has been identified. On the other hand, linkage analysis of southern Italy cases using sets of microsatellite markers identified the HEMPAS causative gene (CDAN2) in chromosome 20q11.2 (3). The nature of CDAN2 is currently unknown. A question remains as to how a defect in a gene encoding a protein other than a glycosylation enzyme results in the disruption of N-glycan synthesis at the GnT-II and MII steps. One possibility is that a defect exists in a Golgi apparatus or cytoplasmic protein regulating membrane trafficking and that this defect affects glycosylation. Biochemical similarities among HEMPAS patients suggest that heterogeneous gene products affect the same biochemical pathway. MII gene knockout mouse: an animal model of HEMPAS A mutant mouse in which the MII gene is inactivated by homologous recombination has been created (4). MII null mice appear normal at birth and grow to adulthood. There are no obvious deformities or life-threatening defects in these mice. MII null mice, however, show various signs of anemia and remarkable splenomegaly. Lectin blots of erythrocyte membrane proteins confirmed a lack of complex type N-glycans in MII null mice, presumably due to the shift of N-glycan synthesis to high mannose type and hybrid type oligosaccharides. Increase of the lacto-series glycolipids, characteristic to HEMPAS erythrocytes, was also seen in the MII null mice. Unsolved problems Phenotypes appearing in MII null mice support the link between genetic defects of MII and HEMPAS. However, cases in which the primary defect is found in the MII gene appear to be limited to the HEMPAS patients from England. HEMPAS in many patients from southern Italy is caused by a mutation in the CDAN2 gene on chromosome 20q11. Besides typical HEMPAS cases, variants are known. These variants may be extreme cases caused by severe mutations of the same gene. It is also possible that undefined genetic factors affect HEMPAS, giving variant phenotypes. It is also possible that genes other than MII and CDAN2 are involved in the variants. | |||||||||||||||||
| Michiko N. FukudaÅ@(The Burnham Institute) | |||||||||||||||||
| |||||||||||||||||
| Mar.15, 1999 | |||||||||||||||||
| |||||||||||||||||

