
John Sheehan received his M.Sc. in Materials Science (1970) and Ph.D. in Biophysics (1973) from the University of Bristol, England, having studied the effects of X-ray fiber diffraction on hyaluronan and other glycosaminoglycans. This work spurred an interest in the applications of materials science and physics in biology that he has followed ever since, as he has elucidated the structural roles of carbohydrates and complex glycoconjugates. He has been a visiting scientist at Purdue University (1979) and at the University of Lund in Sweden (1980-82), where he worked with Drs. Dick Heinegard, Lars-Ake Fransson and Ingemar Carlstedt on the structure and properties of proteoglycans and mucins. He has since concentrated on the biochemistry and biophysics of human mucins, especially in lung mucosal protection mechanisms, first at Lancaster University and subsequently as a Wellcome Senior Lecturer and Reader at the University of Manchester. There since 1993, he has focused on the role of water in biology, particularly in the structure and dynamics of polysaccharides and glycoproteins. In early 2002 he will take up the post of Professor of Biochemistry and Biophysics at the University of North Carolina.

After receiving a Physics degree from Edinburgh University in 1994, Andrew Almond entered the School of Biological Sciences at University of Manchester. His Ph.D. thesis centered on the interaction of carbohydrates (especially hyaluronan) with water, both experimentally and theoretically. After obtaining his degree in 1997, he worked as a Wellcome Trust Prize research fellow at the University of Manchester until 1999, expanding his research into other polysaccharides and oligosaccharides. Under the sponsorship of Professor Klaus Bock, he joined the Carlsberg Laboratory in Copenhagen, Denmark, as a Wellcome Trust Prize traveling research fellow. These he worked to design new experimental approaches to study the dynamic conformation of oligosaccharides, using NMR. In 2001, he joined the group headed by Tony Day at Oxford University, to study hyaluronan-protein interactions, with an emphasis on molecular modeling.
Hyaluronan is a simple, linear glycosaminoglycan, generally found at high molecular weight in many animal tissues. In the last 20 years, scientific attitudes have changed from regarding it only as a molecular cotton wool that fills certain extracellular spaces to ones that also view it as a center around which many matrix macromolecules are organized. A great range of biological functions has been imputed to the molecule, and it has importance from numerous physiological, clinical and diagnostic aspects. Its structural simplicity, span in molecular weight range and unique mode of synthesis mark it as a molecule of distinctive evolutionary significance1. To date, no naturally acetylated, sulfated or other modified variants have been discovered; it appears to be ‘perfect’. This, we suggest, together with the properties outlined above, underlies its functional contribution to biology.
Hyaluronan has many roles, some requiring its presence in minute quantities (e.g., as a proteoglycan organizer in cartilage) whereas in others, it is the dominant structural entity (e.g., its presence in vitreous, Wharton's jelly or synovial fluid). Thus, hyaluronan function may vary greatly depending on whether its interactions are predominantly with proteins in tertiary and quaternary organizations or with water and ions making viscous solutions or gels. As biochemists and biologists, we generally take a rather static view of its functions and properties and have difficulty marrying these two worlds together. There is in particular, some debate as to how the solution properties of hyaluronan in the semi-dilute and concentrated regime should be viewed2-4. It is the aim of this article to consider the molecule in terms of its dynamic capabilities and in doing so to propose an explanation for some of its apparently paradoxical behavior.
There are few views of hyaluronan more static than those obtained in the solid state by X-ray diffraction. In the period from 1970 to 1985, a number of X-ray fiber diffraction studies were done on stretched, semi-hydrated fibers and films of different salt forms of hyaluronan. Some 6 different backbone structures were observed, and each of these could be trapped in a variety of packing arrangements. Of these structures, about 8 have been subjected to detailed X-ray refinement. In general, the quality of the data were good, as illustrated by three different diffraction patterns for the potassium salt of hyaluronan (Fig.1). Refinements of some of these structures gave insight into the preferred chain conformations, molecular packing and ion and water coordination5-7. The origin of these states and how they relate to each other experimentally have been summarized by Sheehan et al.8 Hyaluronan chains are generally found in extended 2-, 3- and 4-fold single helical forms. However, in the presence of the water ‘structure-breaking’ ions (NH4, Cs, Rb and K at low pH) antiparallel, intertwining double helices were found9.
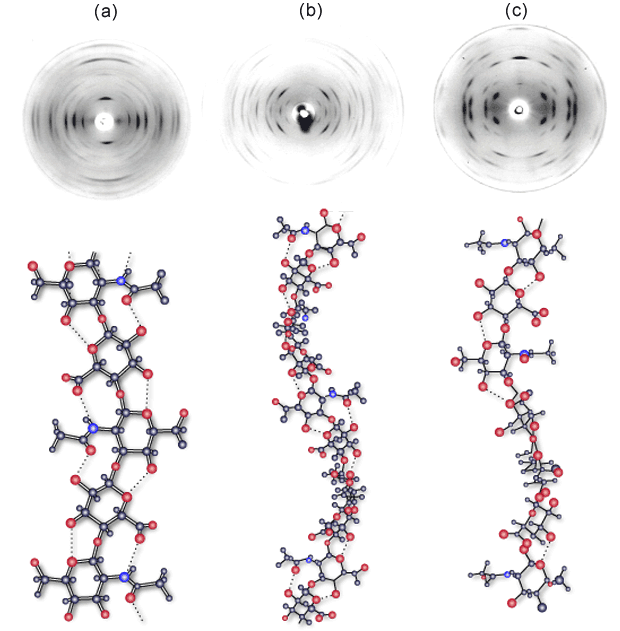
Fig. 1 X-ray fiber diffraction of potassium hyaluronan
Fiber diffraction patterns obtained from potassium hyaluronan at pH 2.0 (a), pH 3-4.5 (b) and pH 5-8 (c). X-ray diffraction in (a) is consistent with an extended 2-fold helix conformation with an axial rise per disaccharide of 0.98 nm, but the structure has never been refined. Diffraction in (b) is consistent with a left - handed 4-fold helix conformation with an axial rise of 0.84 nm per disaccharide. These sinuous chains are organized as two antiparallel double helices in a tetragonal unit cell9. Diffraction in (c) is consistent with an extended left - handed, 4-fold helix of axial rise 0.95 nm per disaccharide. Two of these chains are packed in a tetragonal unit cell6.
How are we to view these data, and what, if anything, do they tell us about the likely state of hyaluronan in solution? Experimentally, these data are achieved by bringing a solution of hyaluronan to the required condition of salt and pH and thereafter drying it down to form a thin film. The film is subsequently cut in strips and stretched at high humidity in order to orientate the polymer chains. Over some time (days to weeks), the chains organize and crystallize. It is clear that this procedure will organize the chains around preferred low energy conformations and packing arrangements. However, the distinctiveness and number of these arrangements was a surprise and suggests that the conformation of the hyaluronan is flexible and very sensitive to the water and cation arrangements around the chains.
Importantly, X-ray fiber diffraction will only ever capture a small sample of the ensemble of conformations available to hyaluronan chains, and it must be borne in mind that the procedure for orientation will itself select against other possible stable conformations. Interestingly, a number of transitions between conformational states could be observed in the solid state. Of particular interest is the effect of calcium, which engenders a change from a 4-fold to 3-fold helical conformation when sodium hyaluronate fibers are washed with calcium chloride8. Distinctive changes have also been observed in viscometric, sedimentation and diffusion properties of sodium hyaluronan solutions when titrating with low amounts of calcium3,10. These data strongly indicate a change in polymer dynamics in the presence of this ion. Treatment of orientated fibers and films with dilute acid solutions also yields a change from 3-fold or 4-fold conformations to a diffraction pattern consistent with an extended 2-fold conformation with an axial rise of 0.98 nm per disaccharide (Fig. 1a). This pattern has never been indexed, probably due to degeneracy of the packing arrangement of the 2-fold chains that leads to the presence of a number of packing arrangements in one fiber. The conformation is not stable above pH 3.0 and is replaced by a variety of 3-fold and 4-fold states, depending on the cation.
A general conclusion from these data is that hyaluronan is highly dynamic in solution and sensitive to the specific ionic conditions, which may engender changes in the bulk solvent, and will certainly make strong demands in terms of ion coordination in a lattice. However, it is interesting to note that in all packing arrangements investigated in detail, there was a strong preference for left-handed helical conformations, and close hydrogen bonding interactions were always between antiparallel chains.
The scaling between molecular weight and size of a polymer in solution is a useful indicator of the ‘average’ shape of that molecule in solution. Hydrodynamic studies on hyaluronan have been pursued over many years. Recently, with its ready availability as a pure reagent, there have been many studies describing its behavior in solution across a wide range of conditions. As alluded to above, the extent of its stiffness is disputed, as are the contribution of intrachain and interchain hydrogen bonds and hydrophobic interactions2-3 to its solution properties. What appears to be agreed is that hyaluronan maintains the status of a viscoelastic solution or paste, depending on the concentration and size of the chains, even up to very high polymer concentrations. It never formally forms solid gels, i.e., hyaluronan solutions are truly viscoelastic and rapidly annealing, unless cross-linking agents of some kind are added. Considering the polymer regularity, as well as the high levels of organization and the close packing demonstrated by the X-ray diffraction data, this is somewhat unexpected. We might intuitively feel that the chains could pack and stack in ways similar to those proposed for agarose, carrageenan and alginate gels and in doing so form similarly rigid gels.
In dilute solution, the agreed description for hyaluronan is that of a stiff random coil, with the chains showing little or no tendency to interact specifically. The persistence length under such conditions is a matter of some contention, with current estimates varying between 4 and 8 nm11. At concentrations in which chains overlap, there is a change in the measured dynamic viscosity. However, recent studies in the semi-dilute regime indicate that the solution is best modeled as a dynamic network of interpenetrating chains3.
There have been some reports in the literature indicating that a number of biological properties of hyaluronan may depend on its size. In our recent work,12 we have attempted to investigate whether there is a change in the hydrodynamic behavior of hyaluronan with size that can be detected in dilute solution. We isolated hyaluronan in a range of sizes from tetrasaccharides up to dodecasaccharides by ion exchange chromatography and from Mr 5,000 up to 100,000 by gel chromatography. Measurements of diffusion coefficients in dilute solution offer a direct measurement of hydrodynamic size and, for the oligosaccharides, were measured by a dispersion technique invented originally by Taylor13 but updated recently12. The method essentially relies upon the broadening effect that a sharp pulse of solute molecules undergo during laminar flow in a narrow bore tube. The method can be made both very sensitive and accurate. The oligosaccharides were modeled as annular cylinders with degrees of extension and mass/length consistent with highly extended structures found in the dynamic modeling and X-ray diffraction. The data were in excellent agreement with the model for oligosaccharides greater than tetrasaccharides (Fig. 2a).
A dynamic light-scattering approach was used to measure the diffusion coefficients of a range of specially fractionated hyaluronans of higher molecular weights and the data combined. These experiments were performed at pH 6.0 (Fig. 2b) (i.e., when the hyaluronan is a polyelectrolyte) and at pH 2.0 (Fig. 2c) (when it is essentially a neutral polymer). At low molecular weight, the relationship between molecular size and weight (obtained from the gradient of the curve) is consistent with highly stiffened, almost rod-like chains (Fig. 2b,c), whereas above Mr 10,000, the behavior changes noticeably and becomes coil-like with increasing molecular weight. This behavior is particularly marked at pH 2.0, when the electrostatic interaction effects are suppressed. Although we expected the chains to behave more as random coils as they got longer, we did not expect to see an identifiable transition zone at rather low molecular weight.
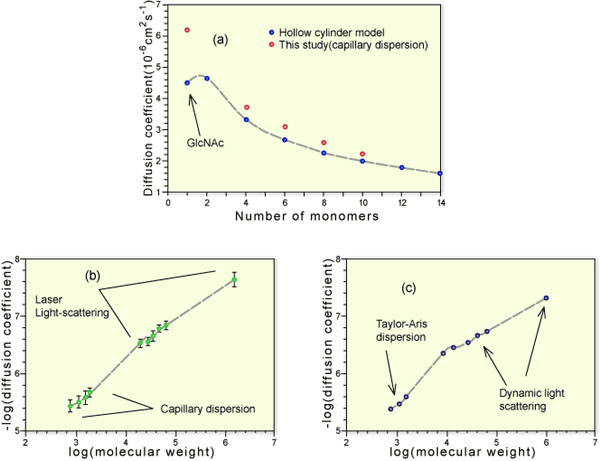
Fig. 2 The relationship between molecular size and molecular weight in hyaluronan
Oligosaccharides of hyaluronan were separated by gel chromatography and ion exchange chromatography. Diffusion coefficients for oligosaccharides were determined by a capillary dispersion method (a) (open circles) and compared with values calculated on the basis of a hollow cylinder model of appropriate mass/length (a) (filled circles). Larger polymers were isolated by gel chromatography and their molecular weights determined by on-line multi-angle light scattering. Their diffusion coefficients were determined by quasi-elastic light scattering and the molecular weight and diffusion data combined in a single log-log plot (b) in 0.2 M NaCl, pH 6.0 (c) in 0.2 M NaCl, pH 2.0. The initial gradient in both cases was consistent with a rod-like structure. However, the gradient beyond Mw 10,000 was consistent with a stiff random coil. This transition was particularly marked in the experiments performed at pH 2.0.
Given that these experiments are performed under dilute conditions, how might such a sharp transition emerge? Taking the most extended length of a disaccharide as 1 nm, the molecular weight dependence suggests that beyond a chain length of 25 nm, there is a change in its properties. We have no proven explanation for the observed change. One speculation under investigation is that the hyaluronan chains below this length cannot form intramolecular segmental interactions to any significant extent, whereas beyond it, the ensemble of their shapes may include such self-interacting forms and thus show, on average, a smaller average size.
As with all biomolecules, the molecular properties of hyaluronan are emergent from its interactions with water. Until recently, there was little intuition as to the nature of these interactions and their persistence as compared with intramolecular interactions. Modern computational approaches can now make realistic dynamic simulations of segments of these molecules in the presence of explicit water molecules. Oligosaccharide NMR studies performed in dimethyl sulfoxide can identify the presence of specific hydrogen bonds14, whereas in water, individual hydrogen bonds have not been observed15. These data suggest that such bonds are in dynamic interplay with the water but cannot give us a precise idea of the time domains involved nor the nature of the averages.
We have undertaken molecular dynamics calculations on di-, tetra- and decasaccharide units in order to build up our intuition of how hyaluronan chains sample their conformational space and what role, if any, intramolecular hydrogen bond interactions play in hyaluronan stability16-18. Figure 3 indicates the dynamic situation, in which a tetrasaccharide is placed in a box containing 1,000 water molecules. It is evident how dominant the activity of the water hydrogen bonds must be against the flexible glycan. We show the data from the decasaccharide simulations to highlight the nature of the analysis we have undertaken.
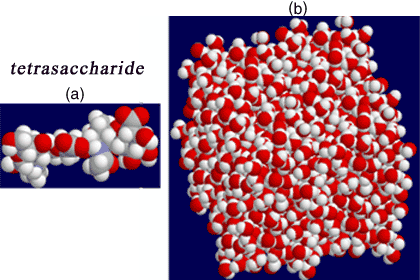
Fig. 3 Molecular dynamics on hyaluronan oligosaccharides
Models of di-, tetra- ( (a) above) and decasaccharide structures have been constructed and placed in an appropriate ‘periodic’ box filled with TIP3P water molecules equilibrated to 300 K (b). Simulations are run for periods up to 5 ns, the coordinates being updated at 1 fs intervals. All spatial coordinates are available for analysis, so that the conformational behavior in time can be examined in detail.
The phi and psi tortional angles were defined as H1-C1-Ox-Cx and C1-Ox-Cx-Hx respectively. Where x=3 at ß1-3 and 4 at ß1-4.
The values of the phi, psi angles defining each glycosidic linkage sampled every 100 fs are shown in Fig. 4. The density of points in such a diagram represents the probability of finding such a conformation, and it can be clearly seen that the ß1-3 linkage is dominated by a single conformation in this simulation, while the ß1-4 linkage is degenerate, i.e., some of the linkages are found in at least two different favorable regions of conformational space. But how is such space being sampled? This question may be answered by documenting the phi, psi angles in time at each linkage. We show this in Fig. 5 for a single ß1-4 linkage. The conformation fluctuates within a single minimum for periods in the range of nanoseconds but undergoes rapid transitions to alternative minima, taking only a few picoseconds to complete the jump.
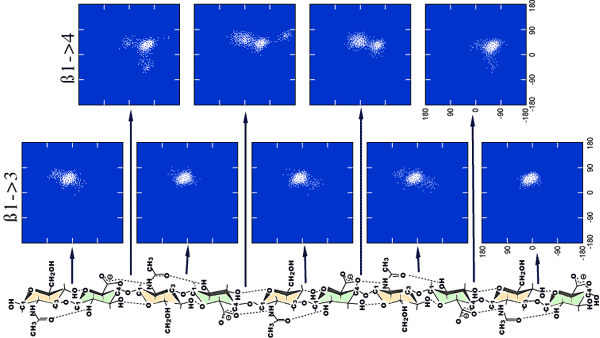
Fig. 4 Output of the glycosidic angles at each linkage of an aqueous simulation of a hyaluronan decasaccharide
The phi/psi angles for each glycosidic linkage have been output every 100 fs throughout a 5 ns simulation. The density of the points reflects the probability of finding the molecule at that conformation and is a measure of the energy. It can be seen that the ß(1-3) linkage is dominated by a single minima, but the dynamic space around the 1-4 glycosidic linkage is more degenerate, and two distinct regions are emergent. There also appear to be ‘end’ effects in the penultimate ß1-4 linkages.
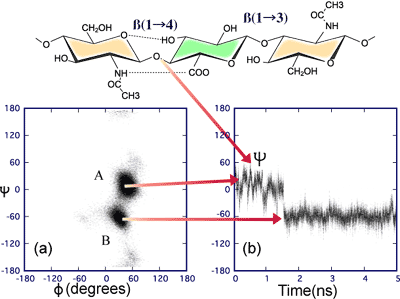
Fig. 5 The time variation of the psi angle at the ß(1-4) linkage
The sampling (100 fs) of the phi, psi angles at a ß1-4 linkage in a 5 ns simulation of a decasaccharide is shown in (a) and indicates the presence of two favored dynamic states. Following the glycosidic angles with time (b) indicates how the molecule jumps from state A to state B by a rapid fluctuation in the psi angle at intervals of the order of nanoseconds. However, the time to make the jump is of the order of picoseconds.
The occurrence of intramolecular hydrogen bonds as the simulation progresses in time may be taken as a measure of how fast the decasaccharide molecule samples conformational space (Fig. 6a). These data indicate that most of the available favorable self-interactions are sampled within 2-3 ns in the simulation. Analysis of these hydrogen bonds indicates that A and B at the ß1-3 linkage are very persistent, while C, D, E and F are very significant at the ß1-4 linkage (Fig. 6b). Little attention has been given to the role of the hydroxymethyl group in hyaluronan conformational studies (E, F), but all our simulations indicate that it plays a key role in hyaluronan dynamics.
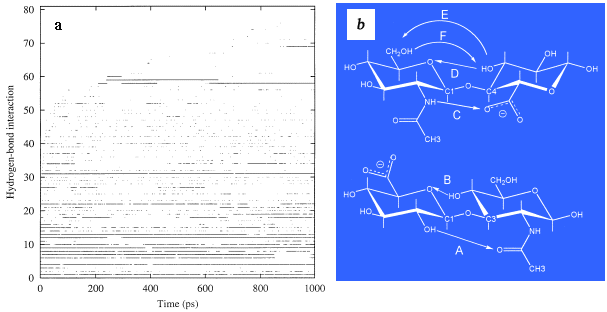
Fig. 6 Intramolecular hydrogen bonds occurring as a function of time
Persistent alignment of hydroxyl groups within the hyaluronan molecule for times in excess of 100 fs is taken as indicative of an intramolecular hydrogen bond. The occurrence of such interactions was documented in time (a) through the simulation and indicates that all such interactions were sampled within a period of 2-3 ns. The most persistent hydrogen bonds are those occurring at the glycosidic linkages and have been highlighted in (b) as A and B across the ß(1-3) linkage and C, D, E and F at the ß(1-4) linkage. The sampling of these bonds in time is shown in Fig. 7.
X-ray diffraction data, taken as a whole, indicate that hyaluronan is strongly sensitive to water and cation environment. The conformations sampled are all quite extended (i.e., the axial rise per disaccharide varies from 0.84 – 0.98 nm), and they show a strong preference for left handed helices. These features are quite consistent with predictions from the molecular dynamics calculations. It is clear that sodium hyaluronan up to a molecular weight of 10,000 can be modeled in solution as a highly extended structure with similar local average dimensions to those obtained from X-ray diffraction analysis and predicted by molecular dynamics. The general tendency towards extended states can also be seen throughout the dynamic simulation (Fig. 7), in which fluctuations in the end-to-end distance tend to decay back to the fully extended state. Thus a number of these low energy states must be consistent with symmetry and chain packing restrictions as well as with ion-water-carbohydrate coordinations. However, we should not infer from the discussion above that hyaluronan maintains specific conformations in solution for long periods. It is clear from the dynamics that hyaluronan is a very rapidly inter-converting set of conformational states and that some of them show marked kinking effects and shortening of the end-to-end distance in extremely rapid time frames.
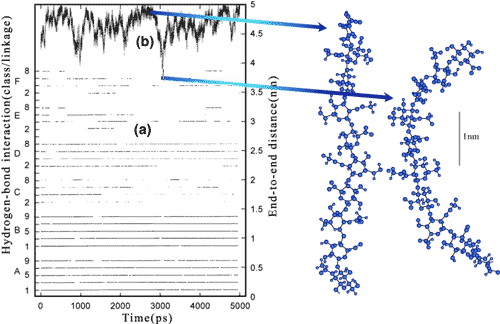
Fig. 7 The variation in the major glycosidic hydrogen bonds and the end to end length of a hyaluronan decasaccharide in time
The hydrogen bonds A, B, C, D at the ß1-4 and E and F at the ß1-3 linkage are shown (a) documented in time. A and B are particularly persistent, whereas there is a rapid exchange between C and D. The fully extended length (b) of a hyaluronan decasaccharide is 4.95 nm, and though the molecule has a tendency to move toward this state, there are rapid fluctuations toward more contracted states. On the right are examples of extended (c) and contracted (d) conformations sampled during the simulation.
The question arises as to how we should think of the role of intramolecular hydrogen bonds versus the formation of hydrogen bonds with the surrounding water molecules. Extended conformations are certainly consistent with the presence of intramolecular hydrogen bonds such as those documented (i.e., A, B, C, D, E, and F), all of which are available to an ensemble of extended conformations. Should we think of hydrogen bonds as ‘stiffening’ the molecule? Our answer would be, probably not. In general, there is little difference in H-bond energy whether the molecule is hydrogen bonded to itself or to water molecules. We have shown previously by detailed analysis of simulations that the water molecules that slow down the dynamics of the molecule and promote intramolecular hydrogen bonding, but paradoxically they also facilitate rapid conversion between conformational states. These rapid jumps between states are catalyzed via water molecules that occasionally get trapped into environments around the glycosidic linkage and transfer energy to the macromolecule. An immobilized water molecule in contact with a dynamic ensemble of water molecules (Fig. 3) is strongly unfavored entropically due to the ordering it imposes in both the biomolecule and the water molecules immediately adjacent to it. Thus, on average, the hyaluronan is pushed towards a state that is consistent with a minimum of disturbances of the hydroxyl, carboxyl and N-acetamido side groups on the dynamic water structure. Such states in all classes of biomolecules are often observed to be consistent with the presence of intramolecular hydrogen bonds, which of course, ‘wrap up’ the electrostatics. From this viewpoint we prefer to argue that many of the extended conformations of hyaluronan are strongly consistent with maximum entropy for hyaluronan plus water.
As noted above, the detailed nature of the dynamics is very interesting. Hyaluronan, though it has a tendency toward extended states, actually explores an ensemble of states that can interchange on the nanosecond timescale, and many of these states can kink the molecule to a considerable degree, as highlighted in Fig. 7, even in short oligosaccharides. The dynamics of the ß1-4 linkage, in particular, indicate that it can rapidly exchange between distinct states that make the molecule conformationally restless. It is our speculation that this dynamic restlessness may underpin the free solution properties of the molecule. In particular, it prevents the formation of long-lived static associations between chains, thus maintaining viscoelastic properties of hyaluronan, and opposes the formation of static networks even at high concentrations. The rapid dynamic conformational interconversion rate will also have important consequences for the dynamic and extensional flow properties of hyaluronan solutions and may underlie many of its important physiological characteristics. The work of Coleman et al. 19, indicates the important role that hyaluronan plays in controlling the pressure/flow-induced osmotic flow across the synovium, while its energy-absorbing properties under extensional flow may be vital for energy dissipation under the rapid dynamic shock associated with our major joints. We would also conjecture that the dynamic properties we have described would make it a perfect ‘space’ colonizing molecule. The nanosecond dynamics of hyaluronan shape change would allow it to fill voids, adjust to surfaces and, when perturbed significantly from its preferred extended states, impose a counteracting force. Such behavior may be important for cell mobility during wound healing and development.
We have stressed the functional importance of hyaluronan dynamics above. However, there are other aspects of its behavior where the importance of its self-similarity and flexibility should be stressed. These attributes make it perfect for building up stable structural complexes based upon cooperative interactions with proteins. It is clear that in many circumstances in the extracellular matrix, different hyaluronan-protein fibrillar complexes are to be found in close proximity20. The evidence suggests that these complexes are generally created external to the cell, and thus, we might expect by the laws of mass action, for them to be somewhat heterogeneous. This is not observed, possibly because hyaluronan provides many differently shaped substrata onto which different proteins can binda. For example, different proteins can draw hyaluronan into a variety of coiling or extended shapes, all of similar energy. These speculations are in the course of being tested by a number of groups, and more insight will soon be forthcoming.
a See review by Day in this series.
We have offered here an alternative vision of how one might view the microscopic behavior of hyaluronan. This vision is based on the surprising proposal (at least to us) that the basic observable properties of solutions of this simple molecule are emergent from the remote picosecond time scale (10-12 sec). It sits on that interesting knife-edge between order and chaos from which so much functionality is emergent. It is currently not possible to make direct measurements on macromolecules on these time scales, but it will become possible to do so in the next few years. Thus, we predict that the understanding solution properties of hyaluronan will continue to tax us intellectually well into this new century.



