Taroh Kinoshita
Research Institute for Microbial Diseases, Osaka University
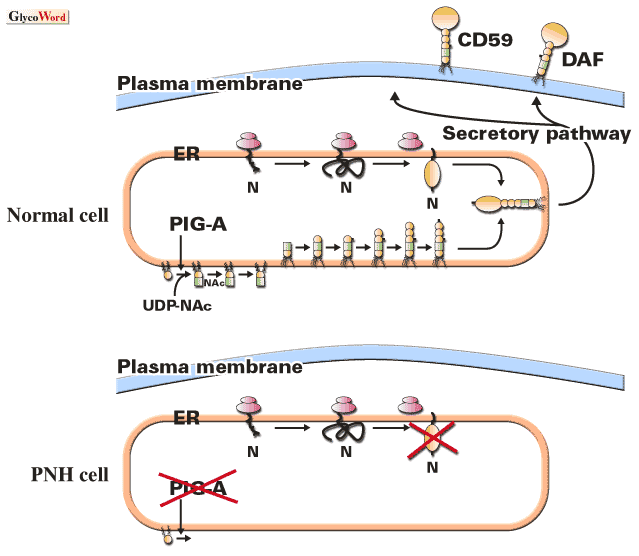
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hematologic disease usually occurring in adults at a frequency of one per one hundred thousand. A major symptom is intravascular hemolysis caused by complement: patients suffer from hemolytic attacks during infections and other events, and have baseline hemolysis that is elevated during sleep, hence the name of the disease. Patients have a clonal population of erythrocytes that are highly sensitive to complement due to deficiency in the surface expression of DAF and CD59 that are GPI-anchored, complement regulatory proteins important for self-protection from complement. The long-living multipotential hematopoietic stem cells harbor an abnormality in the pathway of GPI-anchor biosynthesis that causes defective expression of DAF and CD59.
GPI-anchored proteins are generated in the endoplasmic reticulum by the covalent modification of protein's carboxyl-terminus with a preassembled GPI-anchor, a complex glycolipid. Biosynthesis of GPI-anchor that is initiated by a transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to phosphatidylinositol (PI) consists of at least nine reactions. More than 20 genes are involved in this pathway, 20 of which have been cloned and characterized. Affected cells from patients with PNH are defective in the first step of the pathway. PI:UDP-GlcNAc GlcNAc transferase (GPI-GnT) that mediates this step is a protein complex consisting of at least five gene products, among which PIG-A gene is somatically mutated in affected PNH cells.
The PIG-A gene resides in the X-chromosome at p22.1. One somatic mutation in PIG-A gene, therefore, causes GPI-anchor deficiency in males as well as in females due to the X-chromosome inactivation. Somatic mutations in PIG-A gene have been identified in more than 150 patients with PNH characterized at the molecular level. All other genes involved in biosynthesis and attachment of GPI-anchor would be autosomal. (In fact, other characterized genes are localized in various autosomes.) Two somatic mutations should occur in two alleles of the autosomal gene in the same cell to cause GPI-anchor deficiency. This is a highly unlikely event and should account for uniformity in the responsible gene.
The somatic mutation of PIG-A gene in the hematopoietic stem cell is the first event in pathogenesis of PNH. If the mutant stem cell generates blood cells at a normal level, hemolytic anemia should not occur. But in fact the mutant cells contribute to a major fraction of the patientsユ blood cells as well as CD34-positive bone marrow cells, indicating clonal expansion early in hematopoietic differentiation, the second event in pathogenesis. There are two hypothetical mechanisms of clonal expansion. First, some pathological conditions occur in bone marrow, which in turn selectively suppress or kill normal hematopoietic stem cells, resulting in survival and dominance of the mutant stem cells. Second, some other genetic changes occur in a PIG-A-mutant stem cell, leading to the growth phenotype of the mutant stem cell. Current studies in the field of PNH are focusing on clarifying the mechanisms of clonal expansion.