
Nobuko Hosokawa
Graduated from Kyoto Prefectural University of Medicine in 1984 and trained to work as a clinician for four years. She then obtained her D. Med. Sci. degree from Kyoto Prefectural University of Medicine in 1992. She worked as a visiting fellow in the laboratory of Dr. Carl Wu at the National Cancer Institute, NIH, in 1993. She became an assistant professor at the laboratory of Dr. Kazuhiro Nagata, Chest Disease Research Institute, Kyoto University, in 1994. The institute was reorganized and became Institute for Frontier Medical Sciences, where she is now an associate professor. Her research interest recently is on ER (endoplasmic reticulum) chaperone proteins and the ER quality control mechanism.

Kazuhiro Nagata
Kazuhiro Nagata was born in Shiga in 1947 and graduated from Kyoto University Faculty of Science, Department of Physics. He obtained his Ph.D. (D. Sci.) degree from Kyoto University in 1979. After working for five years in the central research laboratory of Morinaga Milk Company, he joined Professor Yasuo Ichikawa’s group at the Chest Disease Research Institute, Kyoto University, in 1976, working on the differentiation of mouse myeloid leukemic cells. He was appointed lecturer of the Institute in 1979, then joined Kenneth Yamada’s group at the National Cancer Institute, National Institute of Health, Bethesda, MA, and started research on heat shock protein 47 (HSP47), which was later identified as a collagen-specific molecular chaperone. In 1986 he became a professor of the Chest Disease Research Institute which was later reorganized and became the Institute for Frontier Medical Sciences, Kyoto University.
Most of the secretory proteins and membrane proteins are synthesized in the ER (endoplasmic reticulum). ER has a quality control mechanism which discriminates correctly folded proteins from immature or misfolded ones, and retains the latter in the ER (Fig. 1).1,2 Protein folding and oligomer formation are assisted by a group of ER resident proteins known as ER chaperone proteins, and only correctly folded proteins are transported out of the ER to the Golgi apparatus. The folding mechanisms of newly synthesized proteins in the ER are similar to those in the cytoplasm, but ER is different in that the redox potential is biased to oxidization and that most of the proteins synthesized in the ER are cotranslationally modified with N (asparagine)-linked glycans.
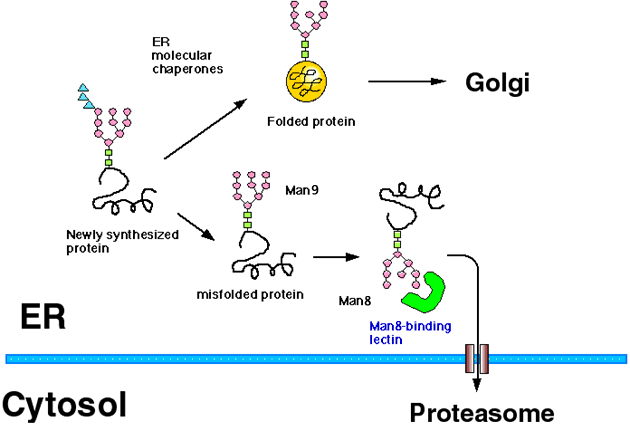
Fig. 1 Scheme of ER quality control and ERAD (modified from Ref. 5)
The ER proteins misfold and sometimes aggregate when cells are exposed to ER stress by the addition of reducing reagents or an inhibitor of glycosylation to cells. Proteins misfold terminally because of premature termination of the protein or by the mutation of even one amino acid residue, such phenomena are observed in some genetic diseases. It has also been reported recently that all the newly synthesized proteins misfold stochastically in part during folding process. ER chaperone proteins such as GRP78/BiP (glucose-regulated protein 78/Immunoglobulin heavy chain binding protein) bind to these misfolded proteins in the ER to prevent aggregation, and support them to refold correctly. Terminally misfolded proteins are destined for intracellular degradation to prevent secretion or transport to its final destinations. Recently, it has been clarified that these misfolded proteins in the ER are degraded by the cytoplasmic proteasome after they are retrotranslocated out of the ER, and this mechanism is known as ERAD (ER-associated degradation) (Fig. 1).3,4 It is now known that not only misfolded proteins but normal proteins can also become the substrate for ERAD. For example, down-regulation of enzymes involved in fatty acid or cholesterol synthesis is mediated by ERAD.
Many of the proteins synthesized in the ER have asparagine-linked Glc3Man9GlcNAc2 glycan added to its consensus sites. In the ER, several enzymes remove and add sugars to the oligosaccharides (Fig. 2A).1,2 Although glycans attached to each glycoprotein are diverse after maturation, the glycan modifications in the ER are uniform. This suggests that the quality control of various glycoproteins in the ER is mediated by a common mechanism through the recognition of the glycan moiety attached to them. Lectin-like molecular chaperones calnexin and calreticulin recognize the Glc1Man9GlcNAc2 oligosaccharide and assist the folding of newly synthesized glycoproteins. Next, the mannose residue from the middle branch of the Man9GlcNAc2 oligosaccharide is removed by ER ![]() -mannosidase I, then glycoproteins bearing Man8GlcNAc2 isomer B (Man8B form) are transported out of the ER to the Golgi apparatus (Fig. 2B).
-mannosidase I, then glycoproteins bearing Man8GlcNAc2 isomer B (Man8B form) are transported out of the ER to the Golgi apparatus (Fig. 2B).
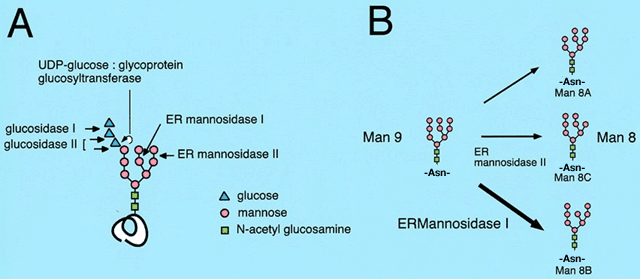
Fig. 2 Structure of N-linked oligosaccharide and its trimming in the ER
The mannose trimming of N-linked glycan also plays an important role in the ERAD of glycoproteins (Fig. 1). In both yeast and human cells, it is reported that the misfolded glycoproteins in the ER are degraded through ERAD only after the glycan is trimmed to the Man8B form, while misfolded proteins stay within the ER when they have the Man9 form of oligosaccharides. Accordingly, it is assumed that there exists a Man8-binding lectin which recognizes misfolded glycoproteins in the Man8B form in the ER and sort them to the ERAD pathway.4
When ER stress is subjected to cells, misfolded proteins accumulate in the ER, which then induces the UPR (unfolded protein response) to cells.6 Consequently, the synthesis of ER chaperone proteins as well as molecules involved in ERAD is upregulated to circumvent the adverse condition.7,8 We have cloned a novel mouse gene which is induced by ER stress using a PCR-based subtraction method.9 This encodes a type II transmembrane protein in the ER consisting of 652 amino acids, and has partial homology with processing α1. α1,2-mannosidase (glycosylhydrolase family 47). We named this gene EDEM (ER degradation enhancing α-mannosidase-like protein), since this gene product was revealed to accelerate the ERAD of glycoproteins. We speculate that EDEM functions as the Man8-binding lectin on the ERAD pathway.
EDEM protein is 18 % identical with human ER α-mannosidase I (ER Man I). Although acidic residues reported to be important for the enzyme activity are conserved, cystein residues which are known to be critical for the enzyme activity are not conserved in EDEM. From the amino acid sequence, we expected EDEM to lack the enzyme activity as a processing α-mannosidase. This was proved experimentally by expressing EDEM as a recombinant protein. We fused EDEM to protein A and transfected it into monkey COS cells. The secreted fusion protein was collected by IgG sepharose beads to prevent the contamination of other cellular mannosidases, and the enzyme activity was measured using 3H-mannose-labeled oligosaccharides. Free 3H-mannose was not released by the incubation with recombinant EDEM protein. Therefore we concluded that EDEM lacks the enzyme activity as a processing α-mannosidase.9
We next examined whether EDEM is involved in the ERAD. We used α1-antitrypsin genetic variant null Hong Kong (NHK) as a model protein, since it is known to be degraded by ERAD.10 HEK 293 cells were transfected with plasmids encoding NHK, and the intracellular degradation of NHK was examined in a pulse-chase experiment using 35S-methionine. The T1/2 of NHK within the cells was approximately two hours, and coexpression of EDEM accelerated the degradation of NHK to approximately one hour of T1/2 (Fig. 3). We have confirmed that NHK was degraded through ERAD in the presence of cotransfected EDEM. Coimmunoprecipitation of EDEM with substrate NHK was detected within the cells. EDEM enhanced the degradation of NHK depending on the extent of mannose trimming from the N-linked glycans. Taken together, we speculate that EDEM is a key molecule which recognizes misfolded glycoproteins and links them to ERAD machinery, and that EDEM is a candidate for the Man8-binding lectin, although actual glycan analysis remains to be carried out.
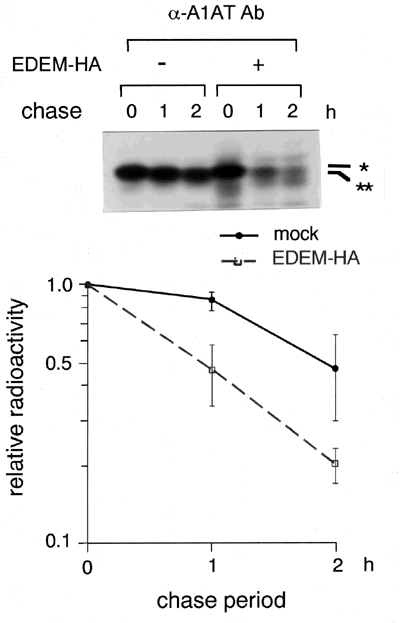
Fig. 3 EDEM accelerates the ERAD of misfolded α1-antitrypsin variant null Hong Kong (NHK). *: Man9 form, **: Man8 form.
EDEM gene is evolutionally conserved and the yeast Saccharomyces cerevisiae has a homologue. It has been reported that the Mnl1p/Htm1p gene, the yeast homologue of mouse EDEM, also accelerates the ERAD of misfolded glycoproteins.11,12
Our working hypothesis on the function of EDEM is schematically shown in Fig. 4. We are now trying to clarify the mechanism of how EDEM works on the recognition and sorting of the misfolded glycoproteins.
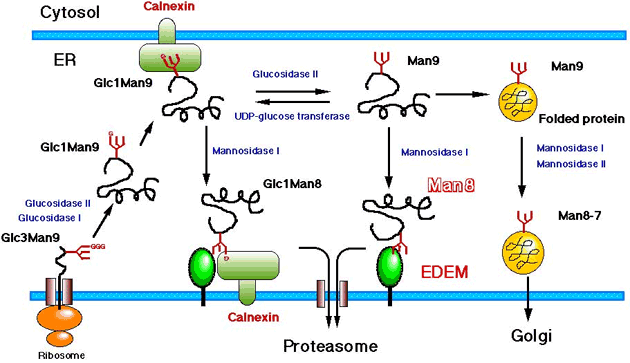
Fig. 4 Scheme of the function of EDEM
As described above, the expression of EDEM is upregulated by ER stress. This is not specific to EDEM, and many of the molecules involved in ERAD are reported to be induced by ER stress.7
ER is an intracellular compartment with high calcium concentration in which proteins are oxidized and modified with N-linked oligosaccharides. ER stress is a condition which accumulates misfolded or unfolded proteins by disturbing these ER circumstances. To circumvent this adverse condition, cells and organisms respond to the ER stress by evoking several adaptation mechanisms.13 First is the transcriptional upregulation of ER chaperone proteins to prevent the unfolded proteins from aggregation and to promote refolding. This signaling pathway from the ER to the nucleus is known as UPR.6 Second, cells attenutate the protein synthesis by phosphorylating eIF-2α to prevent the further overload of newly synthesized proteins in the ER. Third is the induction of the ERAD mechanism described above, and this is regulated at least partly by UPR. The final mechanism of adaptation is apoptosis of cells. Multicellular organisms would survive ER stress by eliminating severely damaged cells.
ERAD system is now clarified to be involved in various cellular mechanisms, and further research is under way in relation to various protein-folding diseases and ER stress.



