
Dr. Jedrzejas received his B.A./M.S. in Physics from Jagellonian University, Krakow, Poland in 1988, a M.S. in Chemistry in 1992 from Cleveland State University/Cleveland Clinic Foundation, and a Ph.D. in Structural Chemistry from Cleveland State University/Cleveland Clinic Foundation in 1993. As a graduate student, he investigated interactions of drug-like molecules with proteins of medicinal interest. His postdoctoral work in microbial pathogenesis was completed in the Department of Microbiology at the University of Alabama at Birmingham School of Medicine and Dentistry. He was appointed an Assistant Professor of Microbiology at the University of Alabama at Birmingham in 1995. In 2001, Dr. Jedrzejas moved to the Children’s Hospital Oakland Research Institute as an Associate Scientist where he directs a structural biology group and continues his studies of the structure and mechanisms of macromolecules using predominantly tools of structural biology with emphasis on X-ray crystallography. His research interests include structural aspects of Gram-positive bacterial pathogens, mainly bacteria-host interactions, with focus on Streptococcus species, and mechanisms of essential processes leading to the formation of bacterial spores, their germination and outgrowth, with an emphasis on Bacillus and Clostridium species. Recently, his laboratory determined crystal structures of hyaluronate lyase enzymes from Streptococcus species and their mechanism of degradation of hyaluronan as a part of a broad effort to understand the role and function of virulence factors of human bacterial pathogens.
Polysaccharide-degrading Enzymes
Microorganisms, such as members of the Streptococcus species, often degrade glycans, including hyaluronan, in order to facilitate invasion of tissues of their host organisms, especially tissues essential for further development and progress of infection.1 The group of enzymes that cleave glycan structures, termed polysaccharide-degrading enzymes, can be divided into two major groups based on their mechanism of action: polysaccharide lyases and polysaccharide hydrolases. In general, polysaccharide lyases utilize the β-elimination process, based on, for example, the proton acceptance and donation (PAD) mechanism or its modifications, to degrade polymeric sugars (described in detail below), and polysaccharide hydrolases utilize a hydrolysis process to accomplish the same cleavage by, for example, direct displacement or double displacement mechanisms.2 Over recent years, the hydrolase mechanism has been studied in greater detail using the three-dimensional structural methods than has the PAD mechanism, which is relatively novel, as it has only recently been fully described.2-5 Both mechanisms, however, have been studied for a long time by biochemical methods. Studies of these two broad mechanisms were significantly aided by the elucidation of three-dimensional structures of several polysaccharide-degrading enzymes. A significant group of these enzymes has a common structural core motif, which consists of a barrel-like structure resembling a horseshoe. This barrel can be described as two sets, or layers of α-helices arranged in a cylindrical, or conical manner one inside the other. The number of helices in each layer varies from 4 to 7 depending on the enzyme.2 One end of such a barrel is usually more open or wider than the other, and this is where the catalytic activity of these sugar-degrading enzymes is usually located. The open end of the barrel has the ability to form an elongated cleft spanning across the whole protein. Such a cleft has appropriate dimensions to accommodate a polysaccharide chain. The cleft surface of these enzymes is usually lined with amino acid residues that are electropositive in their nature, making the surface highly electropositive, a property which is thought to guide these enzymes towards the electronegative groups in their natural polysaccharide substrate chains and to facilitate binding. Once the polysaccharide substrate is bound in the cleft, the catalysis of its degradation can be initiated.4
Hyaluronidases
Hyaluronidases constitute a special group of polysaccharide-degrading enzymes that have the ability to degrade primarily hyaluronan, yielding various lengths of oligosaccharides as the end products. Hyaluronan is one of the components of the extracellular matrices of tissues. Hyaluronan is built from repeating disaccharide units of the chemical structure [→4) GlcUA(β1→3)GlcNAc(β1→]n where GlcNAc is N-acetyl-D-glucosamine and GlcUA is D-glucuronic acid (Fig. 1). Hyaluronan is present in nearly all tissues throughout the vertebrates, and is even present in some bacteria such as Streptococcus zooepidemicus. Hyaluronan isolated from natural sources has enormous size, up to 25,000 disaccharide units or 107 Da.
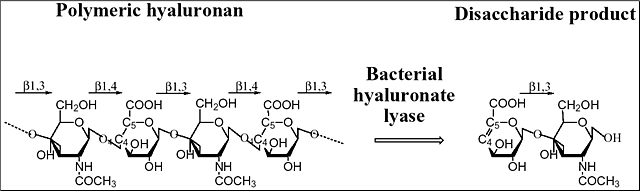
Fig. 1 Structure and degradation of hyaluronan
Hyaluronan is built from alternating units of glucuronic acid (GlcUA) and N-acetylglucosamine (GlcNAc). The representation of the degradation process catalyzed by hyaluronate lyase is shown. The main digestive product of bacterial hyaluronate lyases’ action on hyaluronan is the unsaturated disaccharide unit. The glycosidic linkages between the sugar molecules are also marked.
There are three main classes of hyaluronidases: 1) hyaluronate 4-glycanohydrolase (hyaluronoglucosaminidase), e.g., testicular hyaluronidase; 2) hyaluronate 3-glycanohydrolase, (hyaluronoglucuronidase), e.g., leech hyaluronidase; and 3) hyaluronate lyase, e.g., bacterial hyaluronidase.6 The first two groups of enzymes are hydrolases. Hyaluronate 4-glycanohydrolases cleave β-N-acetyl-hexosamine-(1→4) glycosidic bonds in hyaluronan, chondroitin, and certain chondroitin sulfates yielding even-numbered oligosaccharide units and N-acetylglusosamine at the reducing end of the product. Hyaluronate 3-glycanohydrolases, such as leech hyaluronidase, are specific for the glucuronic linkage in hyaluronan and are inert towards other polysaccharides. Finally, bacterial hyaluronidases are called hyaluronate lyases, and they cleave hyaluronan only at β1,4 glycosidic bonds using the β-elimination process and yield 4,5-unsaturated oligosaccharides of various lengths, often as small as disaccharides. They are different from the two other groups of hyaluronidases, as they do not use hydrolysis in their activity. The two best known hyaluronate lyases, the Streptococcus pneumoniae and S. agalactiae hyaluronate lyases, degrade hyaluronan to disaccharides as their end products, 2-acetamido-2-deoxy-3-O-(β-D-gluco-4-enepyranosyluronic acid)-D-glucose (Δ Di-HA) (Fig. 1). Similarly, the Streptomyces hyalurolyticus hyaluronidase is also an eliminase, but gives mixtures of tetra-, and hexasaccharides as end sizes of hyaluronan degradation. Some hyaluronate lyases such as the S. pneumoniae and S. agalactiae enzymes also cleave certain chondroitins and chondroitin sulfates but only with selected sulfation patterns. The three-dimensional X-ray crystal structures of two hyaluronate lyases from Streptococcus species, one from S. pneumoniae and the other from S. agalactiae, were recently elucidated.2-5 In addition to the native structures, structures of their complexes with substrates were obtained, and biochemical properties including the mechanism of catalysis were analyzed. Based on such analyses, a mechanism of catalysis was proposed.7
The determined three-dimensional X-ray crystal structure of S. pneumoniae hyaluronate lyase (SpnHL) shows a globular protein, which has approximate dimensions of 59 × 59 × 88 Å, and can be divided into two distinct structural domains, α-domain and β-domain, which are connected by a short peptide linker (Fig. 2a).5 The two domains are both spherical and are of similar size. The N-terminal, α-helical domain (α-domain) contains the first 361 amino acid residues of the functional form of the enzyme and contains amino acids from Lys171 to Ser531 of the full length gene from S. pneumoniae. Structurally, this domain is composed of 13 α-helices that are connected by 12 intrahelix residue groups that assume either a coil conformation with two to three residues per turn, or short secondary structures such as β-sheets and 310-helices. The 11 amino acid residue linker from Asp532 to Ser542 covalently connects the α-domain to the β-domain. The C-terminal β-sheet domain, the β-domain, contains 347 residues from Tyr543 to Lys889 arranged in 25 β-strands, which are packed into five antiparallel β-sheets, four short α-helices, and 25 loops connecting the other secondary structure elements.
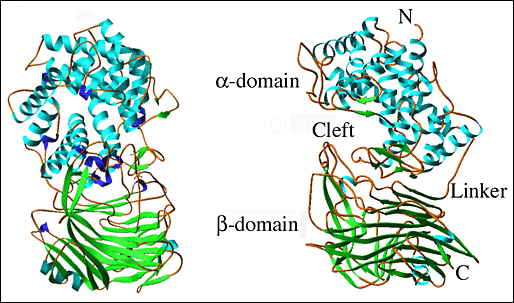
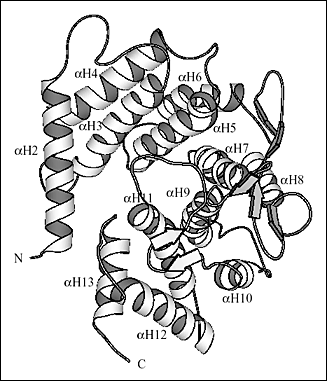
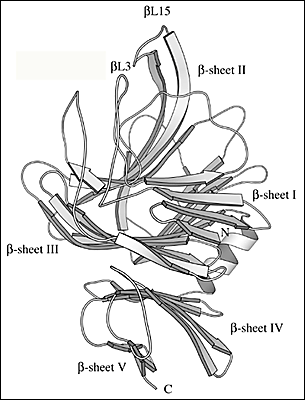
Fig. 2 Three-dimensional structure of S. pneumoniae hyaluronate lyase
(A) Structure of the entire molecule
Both domains of the enzyme are shown, the α-helical domain (α-domain, top) and the β-sheet domain (β-domain, bottom). Perpendicular views of the enzyme are shown. The structure of the enzyme is color coded by the secondary structure elements: α-helices - blue, β-sheets - green, 310-helices - purple, other - brown. The structures are drawn based on the structure coordinates designated 1egu and accessible at the Protein Data Bank (pdb) depository of structures at www.pdb.org.
(B) Details of the three-dimensional structure of α-domain
The end facing the reader is the wider end and the other end is the smaller end of the barrel. The helices were numbered consecutively from αH1 through αH13 starting from the N-terminal. The αH1 helix that blocks the smaller end of the barrel is not shown.
(C) Details of the three-dimensional structure of β-domain
There are five β-sheets in the whole β-domain, and they are labeled β-sheet I through β-sheet V starting from the N-terminus. The two loops that participate in the formation of the cleft spanning the β-domain are βL3 and βL15 (labeled).
Ten of the 13 α-helices of the α-domain are arranged into a barrel-like structure (Fig. 2b). The barrel is incomplete with one terminal end open and wider than the other. The wider end of the barrel faces the β-domain. Also, it is the wider end of the barrel structure together with the loops connecting the adjacent helices that form a cleft in the middle of the SpnHL molecule. It is a prominent feature of the enzyme, and it is where hyaluronan substrate is bound. The cleft traverses the whole α-domain and is relatively straight (Fig 2a and b). It is wide and deep enough to accommodate a polymeric molecule like hyaluronan. The β-domain comprises 24 β-strands that are arranged into five β-sheets (Fig. 2c). The whole β-domain assumes the shape of a four-layered sandwich. Four small α-helix segments are incorporated into several long loops between the β-strands. Finally, there are the two loops from the β-domain, amino acid residues His578-Asn583 and Ile766-Ser771, that participate in the formation of one outer edge of the cleft present in the α-domain. The two domains of the enzyme, in addition to being connected by a peptide linker, interact through the surface interface between them, which is ~3550 square Angstroms in size. This interface is composed of 37 residues from the α-domain and 34 residues from the β-domain. This interface is more hydrophobic in its center and becomes hydrophilic at the outer edge.
The cleft located in the α-domain is approximately 30 Å in length, 10 Å in depth, and 10 Å in width (Fig. 2 a). It is wider at the two distant ends and narrower in the middle. The shortest distance across this cleft is 3.9 Å, and it is between Asn290 from the α-domain and Asn590 of one of the loops of the β-domain. The cleft is the place utilized by the enzyme to bind and to degrade hyaluronan.5 Most of amino acid residues along the cleft are either charged or aromatic. Surface charge distribution shows that one end and the central part of the cleft are highly positively charged and constitute a large positive patch (Fig 3a). Also, three residues, Glu388, Asp398, and Thr400 accumulate at one end of the cleft, which is opposite to the positive patch end. No positively charged residues are present in this area, which makes this end of the cleft electronegative, thus these three residues Glu388, Asp398, and Thr400 form a negative patch (Fig. 3a and b). In addition, there are three hydrophobic residues, Trp291, Trp292, and Phe343, clustered at one side of the cleft, just opposite of the negative patch. This forms a hydrophobic patch (Fig. 3a and b). Finally, three catalytic residues Asn349, His399, and Tyr408 are located in the cleft in its narrowest part, approximately midway between all the patches named above.5
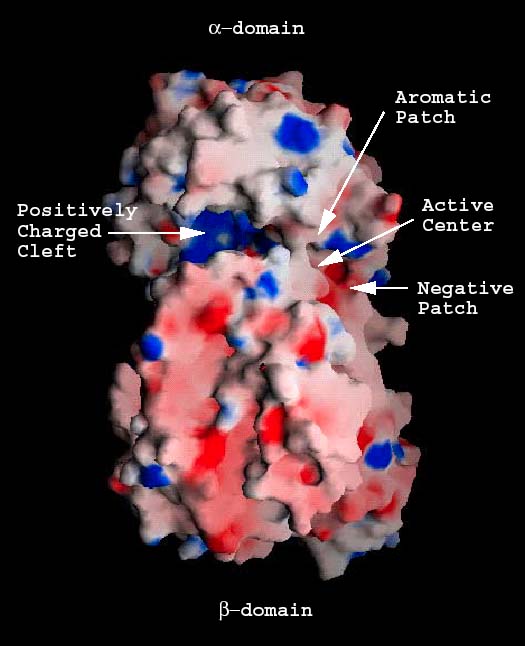
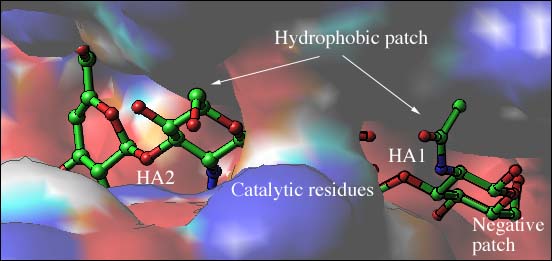
Fig. 3 Electrostatic potential distribution of the S. pneumoniae hyaluronate lyase
(A) Potential distribution for the entire molecule
Potential distribution on the surface of the native molecule is shown. The positively charged cleft (positive patch; blue), the negative patch (red), the aromatic/hydrophobic patch (gray), and the position of the catalytic center residues are identified.
(B) Potential distribution for the binding cleft
A part of carbohydrate binding cleft with a bound hyaluronan tetrasaccharide is depicted with field of view zoomed on the catalytic residues, hydrophobic, and negative patches. The positive potential region is represented in blue and the negative region in red. The hyaluronan substrate is shown as a ball-and-stick model color-coded using standard criteria (carbon atoms in green, oxygen atoms in red, and nitrogen atoms in blue) and disaccharides HA1 and HA2 are shown and labeled. A bridge spans over the catalytic center of the cleft making it closed at this point of the molecule. The catalytic residues are beneath this bridge structure.
Exogenous elements, such as bacteria, degrade hyaluronan utilizing hyaluronate lyase enzymes by the process of β-elimination. In contrast, the endogenous degradation of hyaluronan is performed by the host’s own hyaluronidase enzymes that utilize the hydrolysis mechanism. The exact molecular mechanism of the lyase action was largely unknown until relatively recently when the first structural information on bacterial hyaluronate lyase enzymes was obtained by means of X-ray crystallography. Similar structural information for the endogenous hyaluronidases is not available at present. Therefore, the details of this process are still unclear and are based on the comparison to other polysaccharide degrading enzymes in the class of hydrolases.
The catalytic residues (Asn349, His399, and Tyr408), as well as the residues of the hydrophobic (Trp291, Trp292, and Phe343), and the negative (Glu388, Asp398, and Thr400) patches were clearly identified based upon several studies; including 1) the structure of the native S. pneumoniae hyaluronate lyase,5 2) the structures of complexes with the disaccharide product of degradation, and with tetra- and hexasaccharide hyaluronan units, and 3) site-directed mutation studies.5, 7 The selected mutant enzymes were cloned and isolated. Their specific activities were determined along with kinetic parameters. The correlation between the activity, kinetic properties of the mutants, and the structure clearly identified these residues as being directly involved in various parts of the catalysis.5, 7 Based on the analysis of the above data, a detailed catalytic mechanism was proposed. This mechanism was named the proton acceptance and donation (PAD) mechanism as it involves, first an acceptance of a proton by the enzyme from the hyaluronan substrate, and second a donation of a different proton from the enzyme to the β1,4 linkage glycosidic oxygen. The degradative process consists of five distinctive steps (Fig. 4): (1) In the first step, the enzyme binds the negatively charged hyaluronan substrate in the binding cleft using the predominantly electropositive residues present there, and the substrate/enzyme is precisely positioned for catalysis by the residues constituting the hydrophobic patch; (2) a catalytic step involving primarily the catalytic residues Asn349, His399, Tyr408, and the resulting cleavage of the glycosidic β1,4 bond with the generation of the disaccharide product; (3) hydrogen exchange between the enzyme’s His399 and Tyr408 and the water microenvironment in order to return the enzyme to its natural state ready for the next round of catalysis; (4) an irreversible step of the release of the disaccharide product of degradation utilizing the negative patch in the cleft from Glu388, Asp398, and Thr400; and finally (5) a translocation of the remaining polymeric hyaluronan substrate by one disaccharide unit towards the substrate’s reducing end in order to position the next β1,4 glycosidic bond in the appropriate geometric position ready for the next round of catalysis (Fig. 4).8 These steps are described in more detail below.
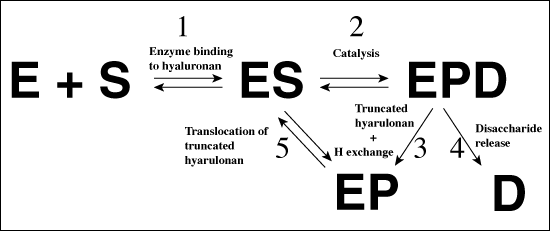
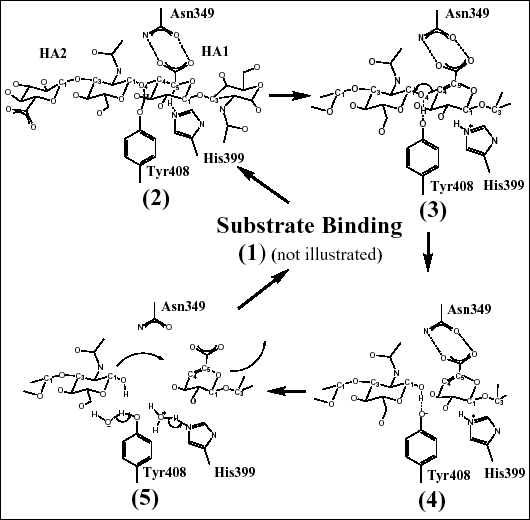
Fig. 4 Schematic diagrams of hyaluronan degradation
(A) Overall mechanism of hyaluronate lyase action
The abbreviations used are: E - enzyme, S - hyaluronan substrate, EPD - enzyme with bound truncated hyaluronan substrate and a disaccharide product, EP - enzyme with bound truncated hyaluronan substrate and disaccharide product released from the active site of the enzyme. See the text for detailed description.
(B) Catalytic mechanism of cleavage of the β1,4 glycosidic bond of hyaluronan
The catalytic residues (and their relative position to the substrate) are shown to illustrate the mechanism of hyaluronan degradation as described in the text. These residues degrade hyaluronan through a five-step proton acceptance and donation mechanism.
Step 1: Enzyme binding to hyaluronan
The enzyme binds to the long chain of hyaluronan utilizing the elongated cleft transversing the α-domain. The cleft has a predominant electropositive character, and its elongated shape perfectly accommodates the substrate chain inside (Fig. 5). Hyaluronan has a predominantly electronegative character, a property that perfectly complements the positive charge of the cleft. The electropositive field generated by the enzyme’s cleft guides the negative substrate into the cleft. As the cleft is primarily built from residues of the α-domain of the enzyme, this domain is the catalytic domain. The β-sheet domain does not seem to play any significant role in this binding process as only two loops from this domain, and predominantly only one residue, Asn580, contribute to the cleft formation (Fig. 5).5, 8 However, the immobilized structure of the enzyme in crystals might not totally reflect the possible changes and the dynamics of this enzyme. As shown by a flexibility analysis, the two domains of the enzyme have a significant degree of mobility with respect to one another.8 They are connected only by the flexible 11-amino acid residue flexible peptide linker, and they also interact through their respective surfaces. Therefore, it is possible that the relative movement of the domains, e.g., α-domain vs. β-domain, could modulate the access of substrate to the catalytic cleft by opening up or closing down the entrance to this cleft (see below for more detailed discussion). After the initial binding of the substrate, hyaluronan is also aligned along the cleft axis by interacting with charged residues lining the cleft surface.5, 8
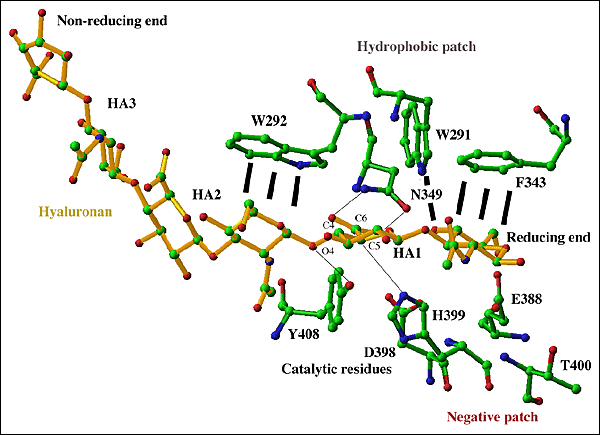
Fig. 5 Active center of S. pneumoniae hyaluronate lyase
The hexasaccharide substrate chain is shown with selected residues of the cleft: catalytic group (Asn349, His399, Phe/Tyr408), hydrophobic patch (Trp291, Trp292, Phe343), and negative patch (Glu388, Asp398, Thr400). Interactions of the catalytic group residues and their targeted substrate atoms are shown by black lines. The substrate chain is shown in yellow, and enzyme residues are shown in standard colors corresponding to atom type. The reducing and non-reducing ends of the substrate are labeled. Both ends of the substrate may extend to include a longer polymeric hyaluronan chain. The β1,4 glycosidic linkage between the HA1 and HA2 positions is cleaved as the result of the process catalyzed by the enzyme.
Based on the structures of the enzyme complexes with its substrates and products, the substrate chain is oriented in the cleft in such a way that the reducing end of the chain is located at the narrower end of the cleft where catalysis is performed. Such unidirectional binding of hyaluronan determines its direction of degradation by the enzyme from the reducing to the non-reducing end (see below). Finally, directly next to the catalytic residues Asn349, His399, and Tyr408 there are three hydrophobic patch residues, Trp292, Trp291, and Phe343 (Fig. 5). These hydrophobic residues interact with the substrate utilizing the hydrophobic character of the sugar rings of the first and the penultimate disaccharide units of the substrates (HA1 and HA2 positions as shown in Fig. 5). Trp292 interacts with the HA2 unit whereas Trp291 and Phe343 interact with the HA1 disaccharide unit. In this way, these three residues precisely position the substrate and its chemical groups in positions that are the most suitable for catalysis (Fig. 4b).
Step 2: Catalytic cleavage of the glycosidic bond
Three residues, Asn349, His399, and Tyr408, were identified as residues directly taking part in the catalysis (Fig. 4b). As visualized by the crystal structures of the complexes with the substrates, Asn349 directly interacts in a bidentate fashion with the carboxylate group of the glucuronate moiety of the HA1 disaccharide. This interaction allows Asn349 to act as a partial electron sink by attracting the electronegative charge of the carboxyl group of HA1 glucuronate away from its C5 carbon. This leads to acidification of the C5 carbon hydrogen (Fig. 4b(2)). As a consequence, His399 is acting as a base and withdraws this hydrogen from C5, and the C5 carbon is rehybridized to a sp2 configuration. In the complex structures, His399 is in direct contact with the C5 carbon of the glucuronate moiety of hyaluronan. At the same time, Tyr408 acts as an acid and donates its OH hydrogen to the glycosidic oxygen O4, leading to cleavage of the β1,4 covalent glycosidic bond between the HA1 and HA2 disaccharides of hyaluronan (Fig. 4b(3)). The C4 carbon that was covalently bonded to the glycosidic oxygen O4 is simultaneously rehybridized to a sp2 configuration, and a double bond is formed between the C4 and C5 carbons of the HA1 disaccharide’s glucuronic acid (Fig. 4b(4)). As a result, a disaccharide product is generated (Fig. 4b).
Step 3: Hydrogen exchange with microenvironment
In the catalytic process described above in step 2, each enzyme molecule gains one hydrogen at His399 and loses one from Tyr408 (Fig. 4b(5)). The hydrogen balance needs to be restored for the enzyme to return to its original state and be ready for the next round of catalysis. There is no evidence in the structures for hydrogen channeling using ordered water molecules (i.e., water molecules seen in the measured electron density of the crystal structure) or other amino acid residues of the enzyme. Therefore, the hydrogens are likely equilibrated using the unstructured water microenvironment present in the enzyme’s cleft.
Step 4: Disaccharide product release
For the next round of catalysis to take place the enzyme also needs to release the generated disaccharide product from the active site. In the proximity of the HA1 position in the cleft (corresponding to the position of the disaccharide product), there are three negative patch residues, Glu388, Asp398, and Thr400 grouped together at the bottom of the cleft surface (Fig. 5). These residues are responsible for the generation of a negative potential in the area of the cleft where the HA1 disaccharide of the electronegative substrate binds (Fig. 5 and 6a). The presence of this patch in this precise location is not coincidental and is important for the function of the enzyme. It is evident that after catalysis when the disaccharide product is cleaved from the polymer, it is directly located in this electrostatic potential. As a consequence, the product of the reaction is irreversibly repelled from the cleft. Such behavior rationalizes the release of the product from the cleft. The electronegative patch facilitates this process and provides a mechanism for freeing up this part of the cleft, thus making the enzyme ready for the next round of catalysis.
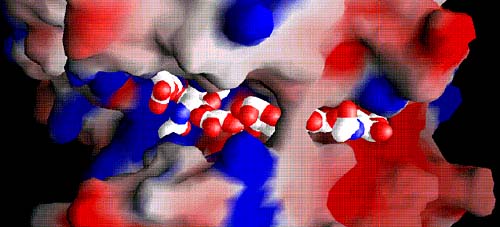
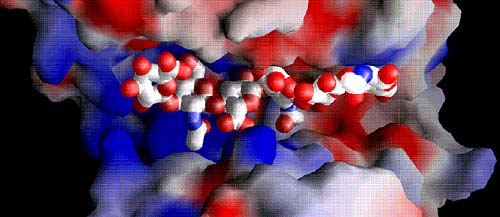
Fig. 6 Electrostatic potential distribution in the entire cleft with the bound hexasaccharide substrate
The positive potential is shown in blue and the negative potential in red. The majority of the cleft is highly positively charged (middle and left side; positive patch), whereas a negative patch is clearly present at the product-releasing end (right side). The hydrophobic patch for the S. pneumoniae enzyme is hidden beneath the bridge spanning over the substrate; the patch in the S. agalactiae enzyme is in the same location. The substrates are shown using van der Waals radii for their atoms; atoms are color-coded using standard colors.
(A) Space filling of hyaluronan bound in the cleft of the S. pneumoniae enzyme
The bridge spanning across the cleft that covers the catalytic center of the enzyme is clearly present. The structure of the cleft region reflects the ‘closed conformation’ of the enzyme.
(B) Space filling of hyaluronan bound in the cleft of S. agalactiae enzyme
The binding cleft is wider than in the pneumococcal enzyme, and no bridge formation spanning the cleft is evident. The structure of the cleft region reflects the ‘open conformation’ of the enzyme.
Step 5: Translocation of the substrate and the processive mode of action of the enzyme
After the release of the disaccharide product, there remains a truncated polymeric hyaluronan in the cleft that can become a substrate for further degradation. One possibility is that the substrate leaves the cleft, and the enzyme binds again to a new or the same hyaluronan molecule using the catalytic cleft (random endolytic or exolytic binding and then cleavage). Alternatively, the molecule already bound in the cleft could stay there and be translocated by one disaccharide unit in the reducing end direction for the next round of catalysis (subsequent processive exolytic binding and cleavage of unsaturated hyaluronan disaccharides by the enzyme). The latter possibility is the case as shown by the biochemical evidence obtained for the S. agalactiae hyaluronate lyase. As the S. pneumoniae enzyme is similar in its sequence, as well as its structural (Fig. 7) and functional properties to the S. agalactiae enzyme,4 it is reasonable to assume that the pneumococcal enzyme behaves in the same way. In this processive mechanism of action of hyaluronate lyase, the enzyme initially randomly binds to hyaluronan and cleaves the polymer into two pieces (random endolytic or exolytic cleavage) (Fig. 4a, step 1-5), of which only the one at the reducing end leaves the cleft (as described for the disaccharide product). The remaining substrate undergoes translocation in the cleft by one disaccharide unit such that the catalysis goes on to generate a disaccharide product, and so on until the chain is fully degraded (exolytic processive degradation) (Fig. 4a, repetitive steps 2-5).
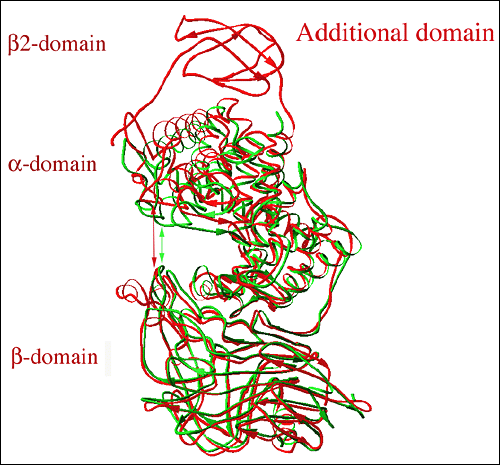
Fig. 7 Structural alignment of three-dimensional structures of hyaluronate lyases from Streptococcus species
The alignment is based on structures for S. pneumoniae (green; pdb code at www.pdb.org: 1egu) with the structure of S. agalactiae hyaluronate lyase (red; pdb code: 1f1s). The S. agalactiae enzyme structure shows the presence of an additional β-sheet domain, β2-domain, at its N-terminus (top part of the figure; labeled). The cleft area in the S. agalactiae enzyme is significantly wider (described as open conformation) than in the pneumococcal enzyme (described as closed conformation). The approximate width of the cleft is marked by arrows, red for the SagHL enzyme structure and green for the SpnHL structure.。
In order to achieve translocation of a polymeric hyaluronan in the cleft, (an essential component of the processive mechanism), several main factors are in place for this enzyme: 1) complementary interface between the enzyme and hyaluronan, and straight shape of the binding cleft that allows for easy substrate translocation or sliding; 2) a correct energetic balance of hyaluronan-lyase interactions allowing the substrate to remain associated with the enzyme after catalysis but also retaining the ability to move along the cleft - a sliding mechanism; 3) a mechanism for the location of the site of catalysis by the hydrophobic patch of the enzyme on the substrate during the translocation/sliding process of the hyaluronan substrate;8, 10 4) a mechanism for the release of the disaccharide product of the reaction to make room for the remaining polymeric hyaluronan to be translocated for further catalysis; and finally 5) the presence of energy to drive the translocation process that originates from energy of charge interactions between the enzyme and the substrate.
Direction of Hyaluronan Degradation
The processive nature of hyaluronan degradation leads to the question of whether this degradation process is performed from the reducing end of the substrate to the non-reducing end or vice versa. Based on the structures of complexes of hyaluronan with the enzyme8 and on the complex structures with the disaccharide product,4, 9 the degradation takes place from the reducing to the non-reducing end of the hyaluronan chain. The currently available structures clearly show that the cleavage of the β1,4 glycosidic bond between HA1 and HA2 disaccharide units of the tetra- and hexasaccharide substrates will produce an unsaturated disaccharide unit at the reducing end of the chain (Fig. 5). The translocation of the substrate in the reducing end direction advances the polymer to the new degradation position in which the HA2 disaccharide takes the place of the cleaved HA1 disaccharide and HA3 that of HA2 (Fig. 4 and 5). In addition, the presence of the negative patch at the reducing end of the bound hyaluronan chain further identifies this reducing end as the releasing end of the substrate.
Flexibility of Hyaluronate Lyase
Computational studies enable correlations between mobility/dynamics of proteins or their segments and specific functions.8 Hyaluronate lyase shows two main types of protein movement; the first movement is a rotation or a twist of the whole α-domain and the part of half of the β-domain that is distant from the cleft; and the second is an opening and closing of the substrate-binding cleft caused by the movement of residues of the negative patch as well as the two loops supporting the edge of the cleft that are from the β-domain (the only part of the β-domain contributing to the cleft formation).
Based on the first major movement, the α-domain acts as a "lid" that opens up or closes down the access to the hyaluronan binding cleft (Fig. 2a). In this selected motion there is no involvement of any residues from the β-domain, even from those that are part of the interface with the α-domain. Also, the residues from the positively charged majority of the binding cleft of the enzyme are especially mobile; the catalytic residues and the negative patch residues do not contribute significantly to this part of the enzyme’s flexibility.3, 8
The second significant motion directly involves modulation of substrate binding in the cleft as well as catalysis by opening up and closing the substrate-binding cleft. The only two loops from the β-domain that are involved in the cleft formation constitute a significant part of this motion. Another significant motion involves the catalytic residues and the residues of the negative patch (Fig. 5). This facilitates catalysis and the release of the product of the degradation.
The structure of S. agalactiae hyaluronate lyase (SagHL) was also recently determined by X-ray crystallography4 and is similar to the structure of the pneumococcal enzyme in the whole architecture as well as in the active site geometry (Fig. 7). The structure contains residues from Ala171 to Ile984 of the full-length gene of the enzyme. In comparison, the crystal structure of SpnHL contains residues from Lys171 to Lys889 of the pneumococcal enzyme gene, which, based on the sequence and structure alignment (Fig. 7), corresponds to Glu249 to Ile984 in SagHL. The α-domain, the peptide linker and the β-domain are also present in SagHL. In addition, the S. agalactiae enzyme has a third domain at its N-terminus, composed from residues Ala171 to Val244, which is primarily built from β-sheet structures, and is termed here β2-domain. The structural superposition of these two streptococcal structures, in addition to explicitly showing the presence of the additional β2-domain, suggests that there likely exists a relative movement between the α-domain and the β-domain as indicated by the flexibility analysis for SpnHL (Fig. 7). The two domains move away from each other, which is accomplished with the help of the linker peptide acting as a hinge. The substrate-binding cleft for this enzyme is therefore wider than that of SpnHL structures by approximately 7 Å (Fig. 6b and 7). As a result, the catalytic residues are also farther apart from each other.
In the SagHL sequence, there are six insertions and no deletions as compared to the SpnHL sequence. A total of twenty residues are inserted. All of them occur at the molecular surface, and five out of the six insertions accumulate specifically around the cleft region. Such behavior might be reminiscent of sequence variations of hyaluronate lyase in order to avoid the host immune system. The cleft region in the SagHL is about 50 Å in length, 10 Å in depth, and ~20 Å in width (~7 Å wider than in SpnHL) (Fig. 6b and 7). The structure cleft of the SagHL enzyme does not have a bridge covering the area of the cleft where catalytic residues are located as is the case for the SpnHL enzyme structure (Fig. 6a and b). The lack of the bridge together with a wider width of the cleft for SagHL likely reflects the flexibility of hyaluronate lyase enzymes.3, 8 The SagHL structure reflects an ‘open conformation’ of the enzyme and the SpnHL structure is in ‘closed conformation’. Residues along the cleft are highly conserved when compared to SpnHL. Based on the comparison to the SpnHL structure, more charged residues are exposed to the cleft surface, and there are more residues from the β-domain that participate in the cleft construction. The active center residues in SagHL are conserved in sequence and in structure in the SpnHL. The catalytic residues of SagHL include Asn429, His479, and Tyr488, the hydrophobic patch residues are Trp371, Trp372, and Phe423, and the negative patch is composed of Glu468, Asp478, and Thr480 (Fig. 6b and 8).4 These residues structurally align very closely with homologous residues of SpnHL. In addition, the catalytic residues are strictly conserved in the sequence of all known bacterial hyaluronate lyases. As expected, mutations of the catalytic residues in SagHL completely inactivate the enzyme.
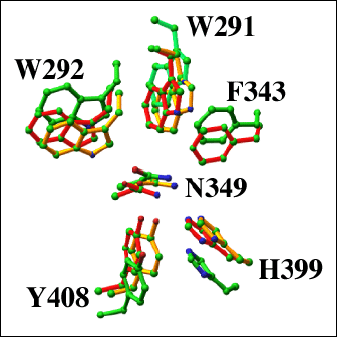
Fig. 8 Structural alignment of the catalytic cleft residues of selected lyase enzymes
The catalytic residues Asn349, His399, and Tyr408 of S. pneumoniae (red; pdb code: 1egu) and S. agalactiae (green; pdb code: 1f1s) hyaluronate lyases, and F. heparinum chondroitin AC lyase (yellow; pdb code: 1cb8) are shown. In addition, the residues responsible for the formation of the hydrophobic patch Trp291, Trp292, and Phe343 are shown.
Activation of Hyaluronate Lyase Enzymes
The analysis of the sequence and the three-dimensional structural information of two hyaluronate lyase enzymes from the Streptococcus genus, S. pneumoniae and S. agalactiae, suggests that the enzymes are synthesized by these two pathogens in their inactive, zymogen form. Then, they are secreted outside of the bacterial cell with activation by cutting of the signal segment, leaving an active form of the enzyme. Both of these enzymes undergo significant degradation before becoming active. At least part of this process is caused by autodegradation. For example, the native, full length SagHL undergoes extensive proteolysis, resulting in a mixture of at least three molecular weight products, 118 kDa, 111 kDa, and 92 kDa forms of the enzyme. The 118 kDa form is enzymatically inactive while the 111 kDa and 92 kDa forms are active. The behavior of SpnHL is similar even though not all of the forms of the enzyme were characterized, unlike the case for the SagHL.
Enzymes with Similar Three-dimensional Structures
Similar barrel structures to that of the hyaluronate lyase catalytic α-domain are observed in other polysaccharide-degrading enzymes. Examples of such enzymes include: Saccharomycopsis fibuligera glucoamylase (Protein Data Bank (PDB) code 1ayx), Aspergillus awamori glucoamylase (PDB code 1dog), Clostridium thermocellum endoglucanase CelA (PDB code 1cem), Sphingomonas species alginate lyase A1-III (PDB code 1qaz), and Flavobacterium heparinum chondroitin AC lyase (PDB code 1cb8) (Fig. 9). The first three enzymes, two glucoamylases and an endoglucanase, are hydrolases, and the S. fibuligera glucoamylase enzyme, for example degrades starch (Fig. 9a). The Sphingomonas species alginate lyase A1-III and F. heparinum chondroitin AC lyase (Fig. 9b) utilize the lyase β-elimination mechanism, and the alginate lyase catalyzes the degradation of alginate in seaweeds and in certain bacteria. The chondroitin AC lyase has the ability to degrade hyaluronan and chondroitin sulfate, likely by the PAD mechanism. It is rather remarkable that the two groups of polysaccharide-degrading enzymes, hydrolases and lyases, use a similar three-dimensional structure to degrade polymeric glycans by such different mechanisms. These enzymes, therefore, probably evolved from a common ancestral enzyme having the ability to bind to and to degrade polysaccharides. The detailed differences in the mechanism of degradation evolved later from the ancestral enzyme and diverged into hydrolases and lyases. The substrate specificity and the precise residues involved in the degradative process, residue conformation, geometry, and organization were likely selected later in the evolutionary development.
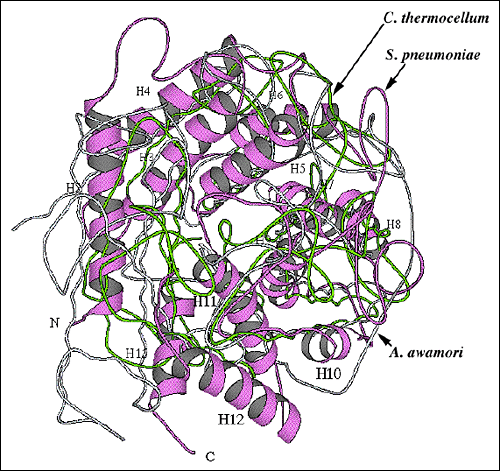
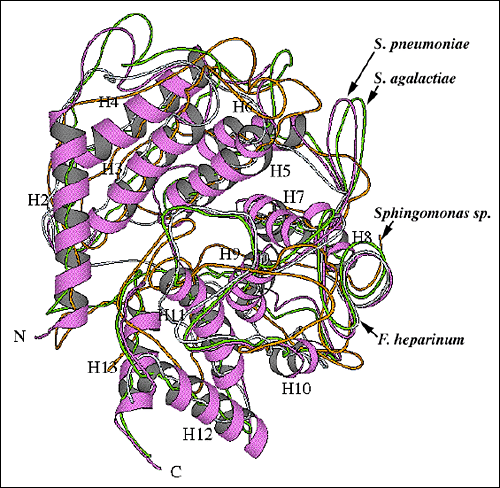
Fig. 9 Structural alignment of overall folds of selected polysaccharide-degrading enzymes.
(A) Alignment of the S. pneumoniae hyaluronate lyase α-domain structure with the enzymes using the hydrolase mechanism
Structural alignment of the S. pneumoniae hyaluronate lyase α-domain (violet ribbon; the same orientation as in Fig. 2b and 9b) with two most similar hydrolases: C. thermocellum endoglucanase CelA (chartreuse; pdb: 1cem) and A. awamori glucoamylase (light blue; pdb: 1dog). The hydrolase enzymes are composed only of the α-helical domain (α-domain). To facilitate the identification of structural elements, the α-helices of the S. pneumoniae hyaluronate lyase α-domain are numbered in the same order as shown in Fig. 2b and 9b.
(B) Alignment of the S. pneumoniae hyaluronate lyase α-domain structure with structures of other lyase enzymes
The alignment of the α-domains of the S. pneumoniae (thicker tracing; the same orientation as in Fig. 2b and 9a) and S. agalactiae (chartreuse) hyaluronate lyases, Flavobacterium heparinum chondroitin AC lyase (blue; pdb: 1cb8), and Sphingomonas species alginate lyase A1-III (orange; pdb: 1qaz) is shown. For the S. pneumoniae hyaluronate lyase, α-helices are shown as ribbons and are numbered as in Fig. 2b and 9a.
Degradation Mechanism
The degradation processes of polysaccharides by polysaccharide lyases were investigated by biochemical as well as structural methods. Recent structural investigations of F. heparinum chondroitin AC lyase and Sphingomonass species alginate lyase A1-III provided some additional insight into this process (Fig. 8). 2, 4 The overall fold of the F. heparinum chondroitin AC lyase is similar to that of S. pneumoniae hyaluronate lyase, consisting of two domains, a α-helical and a β-sheet domain. The α-helical domain supports a similar elongated cleft where the polymeric sugar molecules bind and undergo catalysis. Several scenarios were proposed for the chondroitin AC lyase mechanism, and they are all closely related to the PAD mechanism. This mechanism was proposed to involve residues homologous to Asn349, His399, and Tyr408 of the pneumococcal hyaluronate lyase (Fig. 8).
The current structural information available about hyaluronate lyases includes the structures of the native enzyme from S. pneumoniae and S. agalactiae, the structures of the complexes with the disaccharide product of degradation, and with tetra- and hexasaccharide hyaluronan substrates. The analysis of these structural data together with the body of biochemical evidence allows conclusions concerning the mechanism of catalysis and the functional properties of the lyases. The mechanism of catalysis is well established as proton acceptance and donation. The comprehensive analysis of the entire process of hyaluronan degradation is proposed to include the irreversible release of the disaccharide product from the reducing end of the substrate and subsequent processive cleavage of the adjacent disaccharide. The substrate binding, its precise positioning for catalysis, catalysis itself, the release of the product, and the translocation of the remaining substrate contribute to the entire hyaluronan degradation. Finally, the allosteric behavior of the whole enzyme and its domains as well as that of the hyaluronan binding cleft, including the catalytic area, facilitates the function of hyaluronate lyase.
Acknowlledgements
The readers are encouraged to refer to other sections of this website especially that of Dr. Sakaru Suzuki on ‘Microbial Hyaluronan Lyases’. The studies in the author’s laboratory were supported by grants from the National Institutes of Health (AI 44079) and the Defense Advanced Research Projects Agency (N66001-01-C-8013).



