氏名:井ノ口 仁一
北海道大学大学院 薬学研究科
脂肪細胞から分泌されるTNFαは2型糖尿病におけるインスリン抵抗性を誘導する。 我々は、TNFα刺激で脂肪細胞のガングリオシドGM3合成酵素遺伝子発現が誘導され、増加したガングリオシドGM3はマイクロドメインを介したインスリン代謝性シグナルを抑制することを見い出している。この発見はインスリン抵抗性が基礎疾患として存在する2型糖尿病などの生活習慣病の病態解析に新局面を拓くものである。
インスリンはインスリン受容体(IR)を介して多彩な生理作用を発揮するが、大別すると代謝性シグナルと増殖性シグナルに分類される。インスリンの結合によりIRに内在するチロシンキナーゼ活性が活性化されると、インスリン受容体基質蛋白ファミリー(IRSs; IRS-1, IRS-2, IRS-3)がチロシンリン酸化される。リン酸化されたIRSsはPI-3キナーゼを活性化し、グルコーストランスポーター4(GLUT-4)の細胞質から細胞膜への輸送を促進し、グルコースの取込みが促進される。これは、インスリンの代謝性シグナルの代表例である。一方増殖性シグナルでは、IRの活性化はShcをリン酸化し、RAS-MAPK経路を活性化する(図 1)。
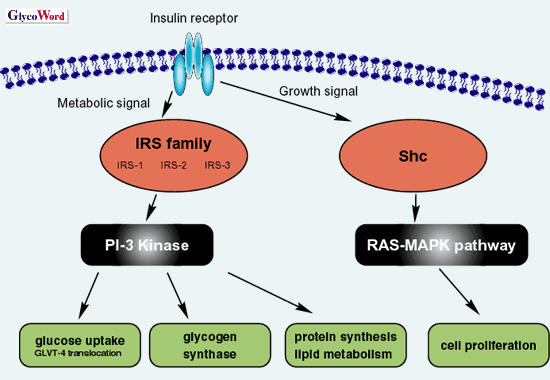
図 1 インスリン受容体と細胞内情報伝達系
1970年に BersonとYalowは、“Decreased ability of cells or tissues to respond to physiological levels of insulin”と定義した。現代社会でインスリン抵抗性は、肥満に起因する2型糖尿病の成因に関連する特徴的病態として重視されているばかりでなく、動脈硬化発症に関わる種々の病態(高脂血症、高血圧症、高インスリン血症など)、即ち生活習慣病の原因となる病態として注目されている。インスリン抵抗性状態では、インスリンの標的組織(肝臓、筋肉、脂肪組織など)において、主としてインスリンの糖・脂質代謝への効果、即ち代謝性シグナルが障害されている。その証拠として、IRS-1の欠損マウスでは、高インスリン血症を示し、インスリン抵抗性が発症する1。
ガングリオシドGM3は、ラット、マウス、ヒトの種を超えて脂肪組織の主要なガングリオシドである。外因性に添加したGM3はEGF受容体2やIR3のリガンド依存性の自己リン酸化を抑制することが示されていた。また、スフィンゴ糖脂質、スフィンゴミエリン、コレステロールなどの複合脂質が集積している細胞膜マイクロドメインにIRが存在していることから4、細胞膜におけるインスリンシグナル伝達においてもGM3が影響を与えている可能性がある。筆者らは、中性脂肪を過剰に蓄積した肥大化脂肪細胞から分泌されるTNFαによって惹起されるインスリン抵抗性はGM3の発現増加によるインスリンとIRS-1間の脱共役が原因であることを見い出した5。ここでは、TNFα刺激によるGM3合成亢進がマイクロドメインの機能異常を引き起こし、インスリンの代謝性シグナルを選択的に抑制している可能性について、我々の最近の結果を中心に紹介する。
2型糖尿病におけるインスリン抵抗性とTNFα
遺伝性肥満とインスリン抵抗性のあるdb/dbマウス、ob/obマウス、Zuckerラットなどの脂肪細胞ではTNFαのmRNAの発現亢進が認められ、また、肥満のヒトでも腹壁皮下脂肪組織のTNFαmRNAおよびTNFαタンパク質が増加している。Zuckerラットに可溶性TNF受容体-IgGキメラタンパク質を投与してTNFαを中和すると、インスリン抵抗性が改善する。また、TNFαを欠損したob/obマウスでは肥満によるインスリン抵抗性が誘導されない。これらの事実から、過剰の中性脂肪が蓄積した肥大化脂肪細胞によるTNFαの産生亢進はインスリン抵抗性を惹起し、2型糖尿病をはじめとする生活習慣病の発症の原因として注目されている。
インシュリン抵抗性惹起物質としてのガングリオシドGM3
マウスの3T3-L1脂肪細胞をIRS-1やGLUT4の発現が抑制が起こらない低濃度TNFα(100 pM)で培養すると、インスリン刺激によるIRのチロシン自己リン酸化の中等度の抑制とIRS-1のチロシンリン酸化の著しい抑制が起こる。この濃度レベルのTNFαで脂肪細胞にインスリン抵抗性状態を誘導するには3日間以上の慢性的な刺激が必要なことから、我々はTNFαによってインスリンシグナルの未知の阻害物質の発現が増加する可能性を推定した。
ガングリオシドGM3は、ヒトを含む種々の動物の脂肪組織における主要なスフィンゴ糖脂質である。そこで、TNFα刺激による3T3-L1脂肪細胞のGM3の発現を経時的に検討したところ、TNFα処理時間に依存したGM3含量の増加が認められ、このGM3の発現増加はGM3合成酵素(SAT-I)遺伝子発現の亢進にもとづくものであった5。また、Zuckerラットおよびob/obマウスの脂肪組織ではleanに比べ著しいSAT-1遺伝子の発現亢進が認められたことから、ヒトの2型糖尿病においてもGM3の発現が増加している可能性が高い。
GM3のインスリンシグナルへの影響を明らかにするために、グルコシルセラミド合成酵素の特異的阻害剤D-PDMP (Glycoword GL-A6を参照)を用いた検討を行った。D-PDMPによりTNF刺激で増加したGM3の発現を抑制するとIRS-1のチロシンリン酸化の抑制がほぼ解除された。この結果から、GM3の増加はIR-IRS-1シグナルを脱共役していることが判明した5。この結果を支持するものとして、SAT-I遺伝子ノックアウトマウスではインスリン刺激に伴うIRの自己リン酸化が亢進していることをNIHのProiaらは見い出している6。
マイクロドメインにおけるインシュリンシグナルの制御
TNFα処理脂肪細胞におけるIRおよびIRS-1の細胞内動態の変化に検討したところ、インシュリン刺激に伴うIRのエンドソーム画分へのインターナリゼーションおよびIRS-1のエンドソームへの集積が正常脂肪細胞と比較して著しく抑制され、IRS-1とIRとの細胞内でのシグナロソーム形成が抑制されていることが示唆された。IRのマイクロドメインへの局在を検討したところ,正常脂肪細胞で認められたIRのマイクロドメインへの局在はTNFα処理脂肪細胞ではほとんど認められず高比重画分に存在が変化していた。興味あることに、TNFα処理脂肪細胞ではインスリン刺激によるMAPK活性化は全く変化しなかった。この事実は、2型糖尿病におけるインスリン抵抗性の病態にはマイクロドメインの異常、即ち、IRのマイクロドメインからの消失が、インスリンの代謝性シグナルの選択的抑制の原因であることを強く示唆している(図 2, 3)。
今後、インスリン抵抗性病態におけるマイクロドメインの機能の変化を、プロテオームおよびメタボローム解析、さらには構造生物学的手法によって明らかにし、ガングリオシドGM3の病態生理学的意義の根本的解明を目指したい。
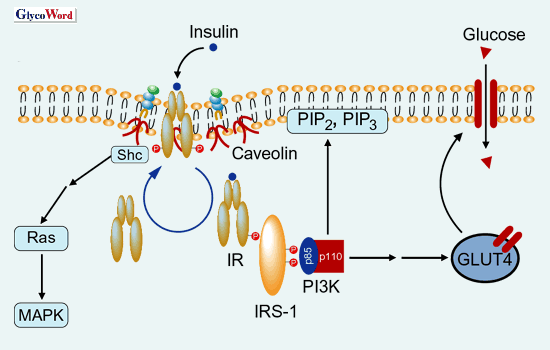
図 2 マイクロドメインを介した正常なインスリンシグナル伝達
インシュリンがIRに結合するとIRの自己リン酸化に続いてIRのエンドサイトーシスが起こり、細胞内のエンドソームにおいてIRSとの会合およびIRSのチロシンリン酸化が数分以内に起こる。チロシンリン酸化を受けたIRSはPI-3キナーゼのPTBドメインと結合することによってPI-3キナーゼが活性化する。このIRS・PI-3キナーゼ複合体はシグナロソームを形成しGLUT-4の細胞質から細胞膜への輸送を促進し、グルコースの取込みが促進される。
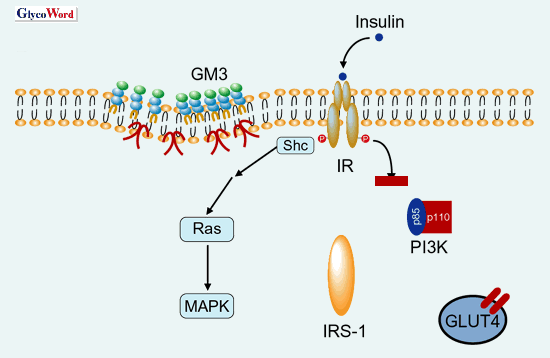
図 3
インスリン抵抗性状態においては、GM3の発現増加に伴ってIRがマイクロドメインから解離し、IRS-1を介するインスリンの代謝性シグナル伝達が選択的に抑制を受ける。一方、Shcのリン酸化に伴うRas-MAPK経路(増殖性シグナル)は変化しない。



