
Dr. Stern obtained his B.A. in 1957 from Harvard College. He worked in the laboratories of Drs. Edmond Fisher and Edwin Krebs as a medical student at the University of Washington, Seattle, where he received the M.D. degree in 1962. He was a postdoctoral fellow at the Weizmann Institute of Science, Rehovoth, Israel, in the laboratory of Dr. Uriel Littauer and then was a Research Scientist at the National Institute of Dental Research, National Institutes of Health, Bethesda, Maryland. Since his move to the University of California, San Francisco in 1977, he has focused on the biology of hyaluronan and the hyaluronidases. He is also a board-certified Anatomic Pathologist and participates in the teaching and diagnostic services of the Department of Pathology.

Dr. Csóka obtained his B.S. degree in Genetics from the University of Newcastle and an M.S. in Molecular Pathology from the University of Leicester, United Kingdom. He received his Ph.D. in Cell and Molecular Biology in September 1998, from the Medical University of Debrecen, Hungary. He is currently a visiting scientist in the laboratory of Dr. Robert Stern in San Francisco. In addition to hyaluronan and hyaluronidase, his interests include the process of aging and the biology of stem cells.
The turnover of hyaluronan in the mammalian body is surprisingly high for an extracellular matrix (ECM) component. The enzymes involved, the hyaluronidases, are broadly distributed. They possess varied substrate specificities and a wide range of pH optima.1-3 Hyaluronidase activity was first identified as a "spreading factor" in extracts from mammalian testes. The term "hyaluronidase" was introduced by Karl Meyer in 1940 to denote the enzymes that degrade hyaluronan. The different types of hyaluronidases were originally classified into three distinct classes by him, a scheme based on biochemical analysis of the enzymes and their reaction products. With the advent of genetic data, we now know that Meyer's classification scheme was remarkably accurate. He identified the three principal types of hyaluronidases:
1) Mammalian-type hyaluronidases (EC 3.2.1.35) are endo-beta-N-acetylhexosaminidases with tetrasaccharides and hexasaccharides as the major endproducts. They have both hydrolytic and transglycosidase activitiesa and can degrade hyaluronan and chondroitin sulfates (CS), specifically C4-S and C6-S, as well as, to a small extent, dermatan sulfate (DS).
2) Bacterial hyaluronidases (EC 4.2.99.1) degrade hyaluronan and, and to various extents, CS and DS. They are endo-beta-N-acetylhexosaminidases that operate by a beta elimination reaction that yields primarily disaccharide end products.b
3) Hyaluronidases (EC 3.2.1.36) from leeches, other parasites, and crustaceans are endo-beta-glucuronidases that generate tetrasaccharide and hexasaccharide end-products.
a Hyaluronan and chondroitin sulfate often occur together in nature, and they also have the ability to bind to each other. The transglycosylation reaction of mammalian hyaluronidases has the ability to generate hybrid molecules in vitro between the two glyosaminoglycan chains. Whether these are generated in vivo, and whether these hybrid molecules might possess some biological activity, has not been investigated.
However, some hyaluronidases do have absolute specificity for hyaluronan such as those from the bacteria Pneumococcus and the mold Streptomyces.
b See article by S. Suzuki in this series.
In vertebrate tissues, the degradation of hyaluronan actually occurs by the concerted action of three enzymes: a hyaluronidase and two exoglycosidases that remove sugars sequentially from the non-reducing termini, a beta-glucuronidase and a beta-N-acetyl glucosaminidase. The endolytic cleavage by the hyaluronidase generates increasing numbers of non-reducing termini for the exoglycosidases. The relative contribution of the endo- and exo- cleavage reactions varies from tissue to tissue, and their individual contributions to overall hyaluronan degradation have not been documented.
Hyaluronidase from testicular extracts has long been recognized4 and characterized.5 However, the hyaluronidases from vertebrate somatic tissues, despite their importance, have until now defied explication. They are difficult to purify, are present at exceedingly low concentrations, and have very high but unstable specific activities in the absence of detergents and protease inhibitors. Recent improved detection procedures for hyaluronidase activity6-8 has facilitated their isolation.
The first somatic mammalian hyaluronidase isolated to homogeneity from a mammalian source was Hyal-1, which was obtained from human plasma. It was then cloned, sequenced, and expressed.9 This 57-kDa, acid-active enzyme comprises a single polypeptide chain of 49 kDa with an additional 8 kDa added by post-translational glycosylation. It is approximately 40% identical to the sperm-specific hyaluronidase PH-20. The mouse ortholog was also cloned and expressed,10 and observed to be 73% identical to the human enzyme.
Human urine has long been documented to contain considerable levels of hyaluronidase. An enzyme identical to Hyal-1 occurs in human urine at 100 times the specific activity of that found in plasma. An additional second band of activity, a 45-kDa processed form, is found in urine. It appears as though the larger protein has had approximately 100 amino acids deleted from the carboxy region by two endoproteolytic reactions (Fig. 1), resulting in two polypeptide chains presumably bound together by disulfide linkages.11 The significance of the second band of activity is not known, nor has it been established if the kidneys are the site of this proteolytic processing. We have preliminary evidence that this second form of the enzyme resides in the lysosome.
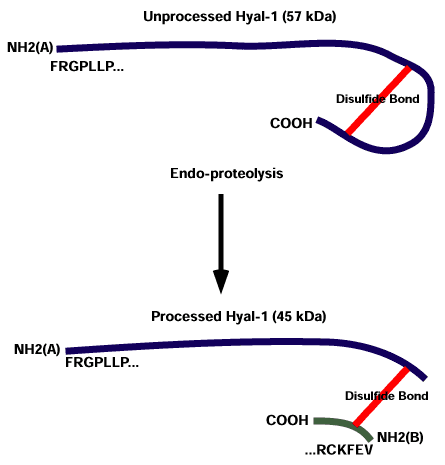
Fig. 1 Diagrammatic representation of the putative endoproteolytic processing of Hyal-1.
Unprocessed Hyal-1 is shown in the upper figure. After endoproteolytic processing, two fragments are generated that produce two separate N-termini. The 22-amino acid-fragment is presumably linked to the rest of the protein by disulfide bonds.
The sequence of the gene for human Hyal-1, denoted HYAL1 by convention, facilitated a screen of the expressed sequence tag (EST) database. This analysis, plus serendipitous discovery of a human sequence contaminating the preliminary Plasmodium falciparum microbial genome database, revealed that the human genome contains six paralogous hyaluronidases sharing about 40% identity with one another. They are grouped into two tightly linked triplets on human chromosomes 3p21.3 and 7q31.3 (Fig. 2). This arrangement may have arisen by an ancient cluster block formation resulting from two duplication events, followed by more recent en masse cluster block-duplication.12
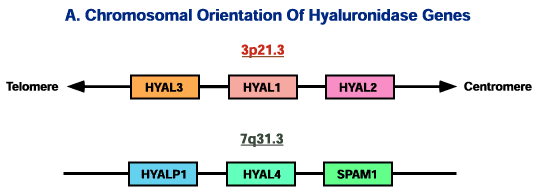

Fig. 2 Demonstration of the chromosomal orientation of the six hyaluronidase genes at their two respective chromosomal sites, and tabulation of their gene products.
The relative gene order has been established for the genes on chromosome 7, but their orientation in relation to the centromere and telomere has not yet been determined. This figure is not drawn to scale.
The Clustal W program13 was used to align the conceptual translation of the cDNAs of five hyaluronidase genes plus the virtual translation of the three "exons" of the hyaluronidase pseudogene, HYALP1 (Fig. 3). Conserved blocks presumably represent regions critical to hyaluronan substrate binding and to enzymatic activity.
Fig. 3
Alignment of the conceptual translation of the cDNA of all five human hyaluronidase genes so far identified, plus the virtual translation of the three "exons" of the hyaluronidase pseudogene using the CLUSTAL W program.13 The sequences for mouse Hyal-1, Hyal-2, and PH-20 are also provided. Identical amino acids are boxed, and similar amino acids are shaded. Conserved blocks, presumably representing regions critical to enzymatic activity, can be seen throughout.
The bee venom enzyme has approximately 30% amino acid identity to PH-20 and Hyal-1 over a stretch of 363 amino acids (Fig. 4). Each of the three sequences contains both a signal sequence and a cleavage site at the amino terminus. PH-20 has an additional cleavage site that releases the enzyme from the transmembrane lipid anchor. The bee venom enzyme lacks the carboxy-terminal third of the vertebrate enzymes, the site of the EGF-like domain in Hyal-1 and the putative cell adhesion domain in PH-20. The transmembrane glycosyl-phosphatidylinositol (GPI) lipid anchor at the carboxy terminus of PH-20 is also present in Hyal-4 (not shown). The EGF-like domain of Hyal-1 has homology to the serrate protein in the fruit fly, Drosophila melanogaster, and to the Xotch protein in the frog, Xenopus laevis. We also observed a weak homology to the human protein Slit2, which is involved in axon guidance.
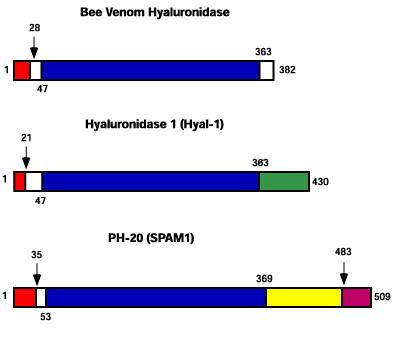
Fig. 4 Diagrammatic representation of putative domains in bee venom hyaluronidase, Hyal-1 (plasma hyaluronidase) and PH-20 (sperm hyaluronidase).
The latter is also known as SPAM1 (sperm adhesion molecule 1). The region of strongest homology between the three enzymes is indicated in blue. The carboxy terminal third of the mammalian enzymes, the region of minimal homology, is absent in the bee venom enzyme. The zona pellucida binding domain, occurs in the carboxy region of PH-20. Similar to PH-20, an EGF-like domain is contained in the carboxy region of Hyal-1. This sequence has homology to serrate protein in the fruit fly, Drosophila melanogaster (52% homology over a span of 66 amino acids), and to Xotch protein in the frog, Xenopus laevis (35% homology over a span of 72 amino acids). Also weak homology is found with the human protein Slit2, which is involved in axon guidance (34% homology over 146 amino acids). Relative polypeptide lengths are drawn to scale.
Crystallographic data available from bee venom hyaluronidase indicates a barrel shape composed of beta/alpha repeats that is common to many glycosidases. The large groove observed on the protein surface of venom hyaluronidase is large enough to accommodate a hexamer of hyaluronan and contains many aromatic and immiscible residues that probably participate in substrate binding and catalysis (Z. Marcovic-Housley, personal communication).
The parallel mouse genes are found on syntenic regions on mouse chromosomes 9F1-F210 and 6A2 (unpublished data). The degree of homology between the pairs of orthologs of human and mouse is much greater than that between the six human paralogs, so the divergence between the paralogs must have occurred long before the divergence of human and mouse, around 80 million years ago. The phylogenetic relationship of mammalian hyaluronidase genes so far identified in human and mouse can be seen in Figure 5. The dendrogram shows that the three genes on chromosome 3 are more closely related to each other than to the two genes and the pseudogene on chromosome 7. It would be interesting to establish when in evolution the gene duplication events that generated the triplet of sequences, and then the en masse duplication, occurred. The genome of Caenorhabditis elegans contains only one hyaluronidase-like sequence with homology to the vertebrate family. Interestingly, Drosophila melanogaster, containing two-thirds the number of genes present in Caenorhabditis elegans, has no hyaluronidase-like sequence.
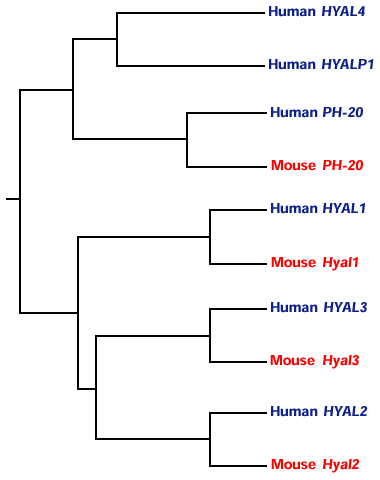
Fig. 5 Phylogeny analysis of six human and four mouse hyaluronidases.
The dendrogram was generated on the Bionavigator web site at http://www.bionavigator.com. Programs used to generate the dendrogram were ClustalW, dnadist, Neighbor, and DrawGram.
All of the hyaluronidase genes have unique tissue-specific expression patterns. The presence of a pseudogene, HYALP1, and the nature of the mutations contained therein suggest that this multi-gene family is still actively continuing to evolve. The genomic structure of all six hyaluronidase sequences can be seen in Figure 6. The genes on chromosome 3 all have a similar genomic structure in terms of intron and exon arrangement, but there is little conservation between the genes on chromosome 7. Furthermore, these genes are considerably larger than those on chromosome 3 because of their higher percentage of non-coding sequences. The smallest gene is HYAL1 at approximately 3.7 kb, and the largest gene is HYAL4 at over 32 kb, a nearly ten-fold variation in gene size. HYAL1 sometimes retains intron 1 within exon 1 in its mRNA. This does not occur in the other genes.
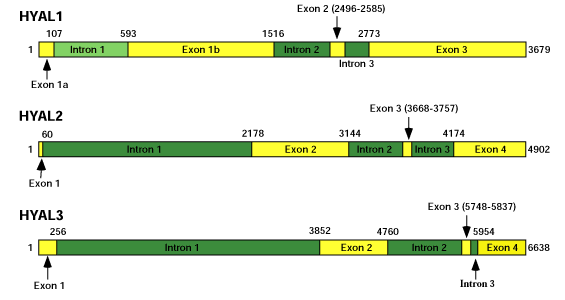
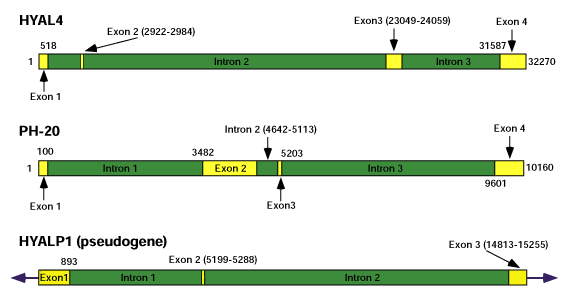
Fig. 6 Genomic structure of the hyaluronidase genes.
Exons are indicated by yellow rectangles and introns are indicated by dark green rectangles. Numbers indicate beginning and end of exonic nucleotides unless otherwise shown. In the HYAL1 mRNA isoform that is not translated into protein (see text), intron 1 is retained within exon 1 (indicated by the light green rectangle.) Exons and introns are drawn to scale for each individual gene, but relative gene sizes are not drawn to scale because of the almost ten-fold spread in size. All of the genes in the hyaluronidase cluster on chromosome 3 have similar exon and intron structures, but this structure is not preserved on chromosome 7.
The gene for the Hyal-1 enzyme was the first somatic hyaluronidase to be identified. Another widely expressed human acid-active hyaluronidase, Hyal-2, is encoded by a gene at an adjacent chromosomal site.14 Hyal-2 has unusual substrate specificity, cleaving high-molecular-weight hyaluronan polymers to intermediate size fragments of approximately 20 kDa. Conceivably, the two hyaluronidases act in concert, and the products of Hyal-2 digestion are further degraded to low-molecular-weight oligosaccharides by the action of Hyal-1. Very little is known about Hyal-3, the third enzyme encoded at the 3p21.3 locus. Strong hybridization expression patterns are found in mammalian testis and bone marrow. These two tissues retain a stem cell-like state for the life of the animal, suggesting that Hyal-3 may be a hyaluronidase first expressed during fetal life. Both PH-20, the sperm-associated hyaluronidase, and Hyal-3 are highly expressed in testis, but whether or not Hyal-3 participates in the process of fertilization is not known.
The protein PH-20 was first identified by monoclonal antibodies present on the acrosomal membrane of sperm. Its homology with bee venom hyaluronidase led to its identification as a hyaluronidase that is essential for both penetration of the hyaluronan-rich cumulus mass that surrounds the ovum and for fertilization. PH-20 is synthesized as a larger polypeptide that is attached to the membrane by a GPI anchor. It is then processed to form a soluble protein composed of two polypeptides linked together by a disulfide bond. It is not known what the relative contributions of the two testicular hyaluronidase enzymes are to the processes of cumulus attachment, penetration, hyaluronan degradation, and fertilization. However, it is intriguing to observe that: 1) the two forms of Hyal-1 in human urine parallel the two forms of PH-20, and 2) both the parent and processed forms are enzymatically active. A human analog of chitinase found in macrophages, which, like hyaluronidase, is an endo-beta-N-acetylglucosaminidase, is processed in a similar manner. The significance of the two forms of all these enzymes is unknown. Since both forms have comparable specific activity, they do not represent the typical zymogen and active enzyme relationship.
A novel hyaluronidase paralog, HYAL4 can be identified at chromosome 7q31.3. The HYAL4 cDNA is calculated to be 2414 nucleotides in length. We have preliminary evidence that indicates that the Hyal-4 enzyme is a chondroitinase with no activity against hyaluronan (unpublished data). This is the first enzyme that is purely a chondroitinase to be identified in vertebrate tissues. It is not surprising that a chondroitinase activity can be coded for in this gene family, considering that the vertebrate hyaluronidases have the ability to cleave chondroitin sulfate, albeit to a limited extent. Chondroitin sulfate is closely related to hyaluronan, the differences being that the N-acetyl-glucosamine is replaced by N-acetyl-galactosamine. Both polymers have exclusively beta-linkages. There is sulfation of chondroitin sulfate, but the possibility remains that the actual cleavage sites are in non-sulfated or under-sulfated regions of the randomly sulfated polymer.
The third sequence, HYALP1, is a pseudogene because multiple deletions exist that cause two premature terminations. The few mutations in the human sequence suggest that HYALP1 may have only recently degenerated into a pseudogene and may be functional in other species. A virtual translation of the exons of HYALP1, illustrating the homology to the other hyaluronidases, can be seen in Figure 6. The amino acids deleted by the mutation in exon 1 and the premature termination in exon 3 are indicated by Xs.
The metabolism of hyaluronan is very active. Rapid increases in hyaluronan levels occur in many clinical situations, including urticaria, the edema associated with wound healing and inflammation, and the organ enlargement that occurs after transplantation. Myocardial damage following infarction is partially due to the pressure necrosis associated with post-traumatic tissue swelling. Circulating levels of hyaluronan rise rapidly in situations such as shock and septicemia, in burn patients. Such increased levels of hyaluronan may be a survival mechanism for the mammalian organism, providing, together with its vast volume of water-of-hydration, an intravascular volume expander that delays circulatory collapse. Hyaluronan and its bound water may represent an entirely separate compartment,not in equilibrium with other compartments functioning purely as a volume expander. The high level of hyaluronan in such settings is a stress response. Holding the rapid turn-over levels of hyaluronan in abeyance, perhaps through hyaluronidase inhibitor-mediated mechanisms, in turn modulated by stress-related cytokines, could provide the mammalian organism a rapid response mechanism for instantaneous elevations in hyaluronan in such dire clinical situations. Preliminary evidence for such a scenario has been obtained (Mio and Stern, unpublished information). The hyaluronidase class of enzymes and their inhibitors may have major therapeutic value for intervention in the aforementioned as well as other clinical situations.
Genetic deficiencies of the different hyaluronidase enzymes could result in clinically distinct syndromes. Indeed, a deficiency of the human plasma hyaluronidase, Hyal-1, has already been reported and is termed Mucopolysaccharidosis IX.16 It is intriguing to speculate that the loss of Hyal-1 activity, as observed in this patient, with only mild clinical features, may be compensated for by the persistence of fetal hyaluronidase, perhaps Hyal-3, a situation that parallels that found in some of the hemoglobinopathies. HYAL4 lies close to a breakpoint in a region of 7q that was found to be inverted in a case of infant "Michelin tire" syndrome, which makes it a positional candidate for this disease.
Hyaluronidases have also been invoked as mechanisms for tumor invasion and metastatic spread. The levels of hyaluronan surrounding tumor cells often correlate with tumor aggressiveness and poor outcome. Overproduction of hyaluronan enhances anchorage-independent growth. Loss of hyaluronidase activity may be one of the several steps required by the multi-step process of tumorigenicity. The HYAL1 gene (also known as LUng CAncer-1, LUCA-1]) maps within a candidate tumor suppressor gene locus defined by homozygous deletions and by functional tumor suppressor activity. Hemizygosity, or loss of one of two alleles, occurs in this region in many oral, head and neck, and lung carcinomas. Mutations in the remaining allele would be required in order for homozygous loss to occur (loss of function in both alleles). For enzymes, being catalytic in nature, it is assumed that loss of only one of the two alleles would not be harmful, and that the remaining allele could supply the cell with sufficient levels of activity. Loss of both alleles, or homozygous loss, would be required to exert an effect. However, no mutations in the HYAL1 region have been reported in carcinomas, despite extensive searches. The classical concept of a tumor suppressor gene (TSG) does not appear to be applicable to this candidate TSG region.
However, we recently identified a lesion that does produce functional silencing of the HYAL1 gene product.17 Neither hyaluronidase enzyme activity nor Hyal-1 protein could be detected in a number of cancer cell lines, even though the mRNA for HYAL1 was present. The unusually large 5' UTR indicated the presence of a retained intron (see Fig. 6). The retained intron blocked translation by preventing the ribosome from binding to the correct initiating methionine codon. It is not known how wide spread such a mode of gene silencing is in carcinomas. However, this finding emphasizes the importance of loss of hyaluronidase in the process of cancer development. Loss of hyaluronidase provides the cancer cell with the hyaluronan-rich environment that stimulates growth, movement, and metastatic spread. These are all components of a successful malignancy. Cancer cells have probably adapted a wide range of mechanisms for silencing unwanted gene products. Apparently, silencing of tumor suppressor genes can occur not only at the genomic DNA level but also, as in the case of HYAL1, at the mRNA level. It should be noted that the 7q31.3 locus also maps to a TSG region.
The turnover of hyaluronan is extraordinarily rapid, but most details of the catabolic mechanisms involved remain elusive. It is not known why the only hyaluronidase in the mammalian circulation is an acid-active enzyme. Is Hyal-1 synthesized by tissues and transported by the blood stream for uptake by other tissues? Are the acid-active hyaluronidases truly lysosomal? If so, why is the pH optimum of such hyaluronidases, pH 3.7, often well below that found in lysosomes, estimated to be pH 4.5? Evidence from patients with Mucolipidoses II and III indicates that Hyal-1 is unusual, and different from most lysosomal enzymes.18 Its activity is not elevated in the plasma of patients with these genetic disorders, indicating either that Hyal-1 is not targeted to lysosomes, or that it is targeted by a mechanism other than the usual phospho-mannosyl recognition pathway. In cultured cells, whether stromal or epithelial, most hyaluronidase activity is quickly secreted into the culture medium and is not retained by the cell layer. Does this reflect the in vivo situation? Are hyaluronidases secreted and then taken up secondarily by cells for delivery to lysosomes? Such a situation is less likely to be recapitulated in vitro by cultured cells, because of the rapidity with which enzymes become diluted in the large volume of medium.
Is receptor-mediated endocytosis involved in hyaluronidase uptake by cells? If so, what is the nature of these receptors? CD44 has been implicated in hyaluronan uptake and degradation. How does CD44 participate in the overall process of hyaluronan uptake and degradation by hyaluronidases? Is CD44, perhaps in one of its multiple variant exon forms, the identity of the receptor for hyaluronidase or for the hyaluronan-hyaluronidase complex? The difficulty with such a formulation is that acid-active hyaluronidases do not bind their hyaluronan substrate at neutral pH. Is there perhaps another molecule or co-factor involved that facilitates enzyme-substrate binding at neutral pH?
In skin, which contains 50% of total body hyaluronan, the half-life of hyaluronan is about one day, and even in as seemingly inert a tissue as cartilage, hyaluronan turns over with a half-life of one to three weeks. In the blood stream, the half-life of hyaluronan is two to five minutes. All such catabolism is presumably a result of hyaluronidases. What is the nature of the control mechanisms that orchestrate such vastly different rates of turnover? The hyaluronan of vertebrate organisms can exist in many states, in a variety of sizes, in extracellular forms, free in the circulation, loosely associated with cells and tissues, tightly intercalated within proteoglycan-rich matrices such as that of cartilage, bound by receptors to cell surfaces, or even in several intracellular locations. Superimposed on these many states are the panoply of binding proteins, or hyaladherins, that decorate the hyaluronan molecule. How do mechanisms of catalysis differ among this wide range of physical and chemical states of the hyaluronan substrate? It is unlikely that hyaluronidase activity is retained in vivo in an active form within the extracellular matrix (ECM) where it could cause great havoc. If it is found within the ECM, it may be in an inactive or suppressed form, perhaps bound to an inhibitor. Such a situation would parallel the relationship between the metalloproteinases (MMPs) and the tissue inhibitors of MMPs, (TIMPS) that exert exquisite control over MMP activity.
The controls for modulating levels of hyaluronidase expression are just beginning to be understood. In cultured cells, whether stromal or epithelial, an inverse pattern of hyaluronan and hyaluronidase activity occurs. As sparsely plated cells grow and begin to fill the dish, hyaluronan levels are high, and acid-active hyaluronidase activity is barely detectable. When cells become contact inhibited and cease proliferating, hyaluronidase activity rises sharply, and levels of hyaluronan deposition fall rapidly.19 Such a scenario is not observed in transformed cells. Keratinocytes obtained from human skin can be made to differentiate in culture when the calcium levels are increased from 0.05 to 1.30 mM. The medium containing high calcium causes cells to stratify and to begin synthesizing keratins. With differentiation, levels of hyaluronan synthesis and deposition decrease,20 while Hyal-1 production increases up to 25-fold.6 The emerging picture is that undifferentiated, rapidly proliferating cells have high hyaluronan levels, but cells lose their hyaluronan-rich matrix in order for proliferation to slow, and for a program of differentiation to commence. Hyaluronidase activities may be key to mediating such transitions.
Recently, hyaluronidase levels in dermal fibroblasts21 were examined during the process of their differentiation into chondrocytes in a micrcomass culture system.22 We found dramatic increases in Hyal-1 expression with differentiation, as well as modest increases in Hyal-2 and Hyal-3. The process of differentiation in various isolated organ and tissue systems can provide many more opportunities for studying the multiple mechanisms for the modulation of hyaluronidase, as first suggested by Bryan Toole in his classic studies of the role of hyaluronan in embryology.23,c
c See article by B. Toole in this series.
There are many other unanswered questions in the area of hyaluronan catabolism. Why are there two forms of the Hyal-1 enzyme? What is the nature of the protease(s) that perform the endoproteolytic reactions, and where do these cleavage reactions occur? Do the two forms of Hyal-1 parallel the two forms of PH-20, the membrane-bound and the soluble form? Which of the two forms of Hyal-1 is more active in vivo? Why are there two forms found in urine but only one in plasma, when cultured cells can express both? What is the significance of the very high levels of hyaluronidase activity in urine, all of which appears to be Hyal-1? Are there other hyaluronidases yet to be discovered that account for the grams of hyaluronan that are degraded each day in the mammalian body? A 70 kg individual possesses about 15 g of hyaluronan, a third of which turns over daily. The turnover of hyaluronan is obviously tightly controlled by temporal and spatial mechanisms about which we know very little. Perhaps additional hyaluronidases exist, unrelated to the family of six, that have not been identified to date because of the artifacts inherent in in vitro enzyme assays. Clearly, many questions remain concerning the mammalian hyaluronidases.



