
氏名:佐藤 智典
慶應義塾大学理工学部生命情報学科教授
1983年3月九州大学大学院総合理工学研究科修士課程を修了、1983年9月同博士課程を中退し、学位は1990年に京都大学で工学博士を取得。職歴は、1983年10月長崎大学工学部助手、1990年10月京都大学工学部助手、1992年4月東京工業大学生命理工学部助教授、2000年4月慶應義塾大学理工学部助教授を経て、2002年4月より現職。受賞は、1993年日本化学会進歩賞、2010年バイオビジネスコンペJAPANバイオ先端知賞など。研究テーマは、糖鎖プライマーを用いたグライコミクス、多糖を用いたドラッグデリバリーシステム、生体膜モデルを用いた脂質ラフトの形成と認識機能の解析、ファージディスプレイ法による糖鎖関連ペプチドの探索とその機能解析など、糖鎖に関して多様なテーマに取り組んでいる。
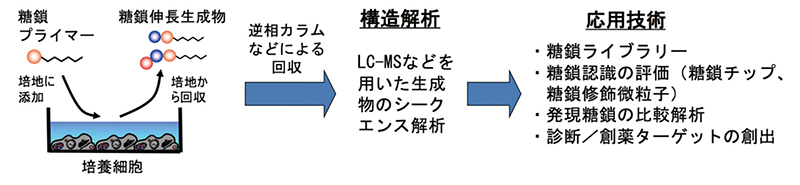
糖鎖プライマーとは、糖鎖の生合成経路の前駆体となると糖誘導体である。糖鎖プライマーを細胞と相互作用させることで、糖鎖生合成経路に従った糖鎖伸長生成物が得られる。糖鎖伸長生成物の構造は質量分析装置(LC-MS)を用いて解析し、糖鎖ライブラリーの構築や糖鎖プロファイリングを行っている。グライコミクスの新たなツールとしての糖鎖プライマー法について紹介する。
細胞に発現する糖鎖は、糖脂質、糖タンパク質、プロテオグリカンなどの多様な複合糖質として細胞表面に提示されており、発生・分化・増殖・遊走性などの多様な細胞機能と密接に関係している。細胞の機能を知るためには、細胞に発現している糖鎖の全体像を理解する必要があり、そのために糖鎖プロファイルの作成が求められている。遺伝子やタンパク質の発現解析は活発に行われており、ゲノミクスやプロテオミクスは飛躍的に発展してきた。一方で、糖鎖の解析は一般的な技術として生命科学の分野で定着しているわけではない。そのために、グライコミクスの研究分野の裾野の広がりは見られていない。そのことを打開して糖鎖研究を発展させるためには、簡便な糖鎖解析手法の研究開発は不可欠である。
グライコミクスには2つの命題がある。一つ目は、細胞に発現している糖鎖の全体像を明らかにすることであり、二つ目は、その糖鎖の役割を解明することである。その二つを達成することができれば、生命科学における糖鎖機能の学術的な意義を多くの研究者にアピールすることが可能になり、疾病における診断や創薬分野にも貢献できる。また、グライコミクスの命題を達成するには、細胞に発現する糖鎖の種類や機能を解明するためのGlycoscienceと、糖鎖やその誘導体を作製してその機能を検証するためのGlycotechnologyの両方が不可欠である。GlycoscienceとGlycotechnologyはそれぞれに高い専門性が必要なので、異なった研究者により実施されてきたが、二つの分野の研究者が連携することで革新的な発展が生まれて来た。その様な中で、筆者は、GlycoscienceとGlycotechnologyの橋渡しをするための技術として、糖鎖プライマー法の開発を行ってきた。その概要について紹介する。
糖鎖プライマーとは、糖転移酵素の基質となる糖鎖生合成の前駆体である。糖鎖プライマーは糖鎖生合成の起点となる糖残基を有しているので、細胞と相互作用することで多様な糖転移酵素により糖鎖プライマーに糖鎖が伸長される。これまでに、糖転移酵素の基質となる糖誘導体が報告されてきた。例えば、p-nitrophenyl β-D-xyloside は糖転移酵素の基質となり1、4-methyl umbelliferyl β-D-xylosideは糖脂質とGAGの合成を阻害することも見出されている2。p-nitorophenyl α-D- GalNAc にはムチン型のO-glycanの糖鎖が伸長していた3。また、peracetylated Xylβ1-6Gal-O-2-naphthol、peracetylated Galβ1-4GlcNAcβ-O-naphthalenemethanolが細胞内での糖鎖伸長反応の基質になること4,5。およびその様な化合物は内在性の糖鎖の生合成の阻害剤になることが報告されている4-7。糖誘導体や生成物などが細胞内に留まることで糖鎖生合成を阻害すると考えられる。
このような研究に対して、本著者は糖鎖プライマーのアグリコン部分にはドデシル基などの炭化水素鎖を用いている(図 1)。炭化水素鎖の長さを最適化することで、細胞内に糖鎖プライマーが取り込まれた後、糖鎖プライマーや糖鎖伸長生成物は細胞内には蓄積されず、大部分は細胞外に分泌されていた8。そのために、糖転移酵素の阻害剤としての活性は低く、糖鎖伸長反応が優先して観察された。アルキル基を有する糖鎖プライマーでは、糖鎖伸長生成物が細胞外に分泌されることで、細胞をホモジナイズすることなく生成物を回収できるので、糖鎖合成という観点でもメリットを有している。これまでに、糖残基としては、ラクトース(Lac)8-11、 N-アセチルグルコサミン(GlcNAc)12、N-アセチルガラクトサミン(GalNAc)、およびキシロース(Xyl)13-16を用いている。GalNAcとXylはO-グリカンの生合成の前駆体となるので、糖残基にSerあるいはThrのアミノ酸残基を連結した糖アミノ酸を糖転移酵素の認識部位として用いている。
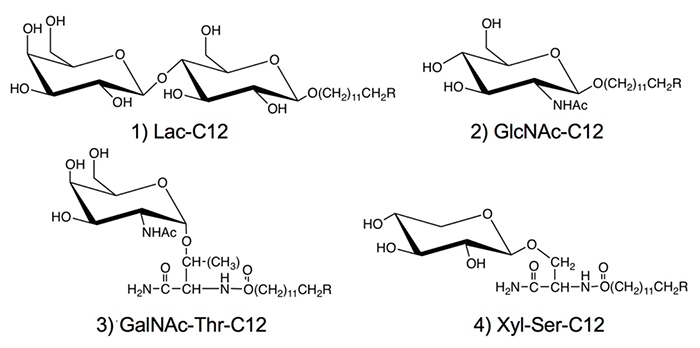
図 1の糖鎖プライマーでは、生成物が細胞外に分泌されるという特性があることから、本著者らは糖鎖ライブラリーを構築することを目指して研究を行ってきた。糖鎖ライブラリーを作るための糖鎖合成では、有機合成や酵素合成が一般的に行われてきた。これに対して、糖鎖プライマー法では、細胞を糖鎖の合成工場として活用することで糖鎖ライブラリーを構築する。そこで、細胞に糖鎖ライブラリーを作らせる手法は、有機合成法と酵素合成法に続く第3の手法といえる。
糖鎖プライマー法により細胞で糖鎖を作るための実験は次のような手順で実施している(図 2)。具体例としては、直径10 cmの培養ディッシュで細胞を培養して、サブコンフルエントになった時点で通常培地を50 µMの糖鎖プライマーを含んだ無血清培地に交換する。2日間培養したのちに、培地中に分泌されている糖鎖伸長生成物を回収する。細胞を破壊することなく生成物を回収できるので、糖鎖プライマーを複数回繰り返して添加・回収することも可能である。回収した培地成分は逆相カラムを用いて脱塩し、生成物を固相抽出する。溶出溶媒の組成を変えることで酸性と中性の生成物に分離することもできる。得られた生成物の構造は質量分析装置により解析する。固相抽出した糖鎖伸長生成物は糖鎖の混合物であるので、LC-MSを用いて糖鎖構造の解析を行う。LCではシリカカラムなどを用いて生成物を分離し、MSではエレクトロスプレーイオン化/イオントラップ型(ESI-IT)や四重極飛行時間型(Q-TOF)などを用いてMS2を測定して配列解析を行う。
LC-MSでの糖鎖解析では、LCによる構造異性体の分離が可能という利点がある。例えばNeuAc-Galでは、α2-3とα2-6結合を区別する必要がある。両者ではLCでの保持時間は異なり、NeuAcα2-3Galでは短い。また、MS2測定で検出されるフラグメントイオンから両者の結合様式の区別も可能である。例えば、NeuAcα2-3Galβ1-4GlcNAc-C12結合ではMS2スペクトルのm/zが823.4と550.2にピークが検出されていたが、NeuAcα2-6Galβ1-4GlcNAc-C12結合ではそのピークが殆ど検出されずそれに代わってm/z 335.9が強く検出された。また、Gal-GlcNAc結合ではβ1-3とβ1-4の結合様式を区別しなくてはならない。LCでの保持時間はGalβ1-3GlcNAcが短く、Galβ1-4GlcNAcでは長い。さらに、Galβ1-4GlcNAcとGalβ1-3GlcNAcのMS2スペクトルにも違いが見られた。以上の様に、LC-MSの測定において、分子量が同じであっても、LCの溶出時間とMS2スペクトルを解析することで構造異性体を簡便に判別できる。LC-MSによる糖鎖構造の同定は有用な手法ではあるが、装置の感度との兼ね合いで存在量の少ない糖鎖では結合様式を決定するのは難しい場合もある。
オリゴ糖の結合様式の区別に加えて、単糖の構造異性体の区別を行うことは、糖鎖の構造解析を難しくしている要因である。例えば、ヘキソースHexではグルコースGlc、ガラクトースGalおよびマンノースManを、またN-アセチルへキソサミンHexNAcではN-アセチルグルコサミンGlcNAcとN-アセチルガラクトサミンGalNAcを区別しなくてはならない場合が多い。単糖の構造異性体の区別は、ガスクロマトグラフ法により行われてきた。糖の構造異性体の区別を質量分析装置で行うことができれば、糖鎖の配列解析の精度の向上になる。そこで、ESI-IT型のMSによる構造異性体の区別を試みた17。
単糖の構造異性体を区別するために、MSの測定条件を検討したところ、溶媒の影響は大きく、酢酸アンモニウムを含有する糖溶液を用いることが有効であった。また、ポジティブイオンモードでのMS2およびMS3のスペクトルを測定する際に、適切なプレカーサーイオンを選ぶことで、糖の種類によりフラグメントイオンの強度比が異なることを見出した(表 1)17。例えば、ヘキソースのGlcとGalの違いは、プレカーサーイオンがm/z198の時にMS2スペクトルのm/z163とm/z180の強度比の違いにより区別できた。GalとManの区別もMS3スペクトルを測定することで可能であった。GlcNAcとGalNAcの違いはMS2スペクトルでは区別することができなかったが、MS3スペクトルで3つのフラグメントイオン(m/z=126,168,186)の強度比を測定することで区別できた。一方、ネガティブイオンモードでは、同位体の区別はできなかった。この様に、イオントラップ型の質量分析装置では、測定条件を選ぶことで構造異性体の分析が可能であった。
単糖の構造異性体の区別は、オリゴ糖を用いた解析の際にも適用できた。その例として、Gal-GlcNAc誘導体での測定を行なった。MS2スペクトルではHexNAcに相当する分子量のフラグメントイオンが検出され、これをプレカーサーイオンとしてMS3スペクトルを測定するとm/z 204のフラグメントイオンが検出された。次に、m/z 204をプレカーサーイオンとしてMS4スペクトルを測定すると、表 1に示す様なGlcNAcに特徴的なフラグメントイオンが検出された。オリゴ糖の配列解析において、GalNAcとGlcNAcを区別するために通常は酵素処理が必要である。その様な実験の煩雑さを回避できることから、MSnスペクトルが測定できるイオントラップ型の質量分析装置は、糖鎖の構造解析に有用であると考えられる。但し、MS4スペクトルの測定では、シグナル強度を得るための十分なサンプル濃度が必要であり、また装置の感度も影響してくる。
| 糖の種類 | MS2スペクトル | MS3スペクトル | ||
| プレカーサーイオン | フラグメントイオンの強度の大小 | プレカーサーイオン | フラグメントイオンの強度の大小 | |
| Glc | m/z 198 [M+NH4]+ | m/z 163 < m/z 180 | m/z 163 | m/z 127 < m/z 145 |
| Gal | m/z 163 < m/z 180 | m/z 127 ≈ m/z 145 | ||
| Man | m/z 127 < m/z 145 | |||
| GlcNAc | m/z 222 [M+H]+ | m/z 186 < m/z 204 | m/z 204 | m/z 126 ≤ m/z 186 m/z 126 > m/z 168 |
| GalNAc | m/z 126 < m/z 186 m/z 126 < m/z 168 |
|||
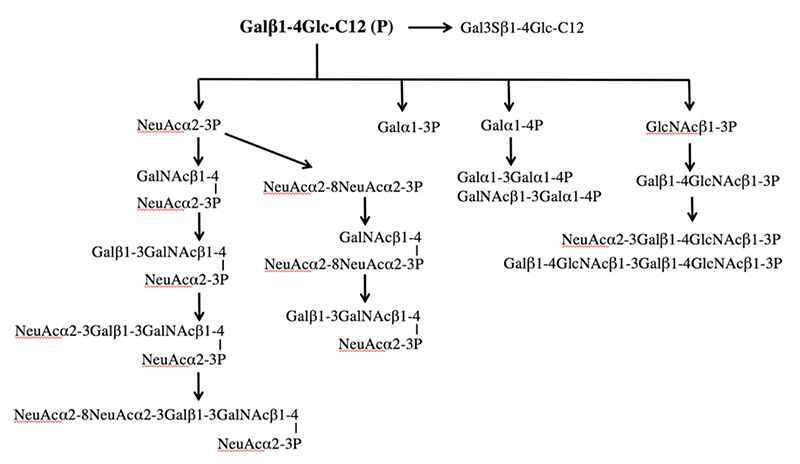
図 1に示す糖鎖プライマーは異なった糖鎖生合成経路での前駆体である。また用いる細胞は株毎に異なった糖鎖生合成経路を発現している。そこで、糖鎖プライマーと細胞との種類の組み合わせにより、多様な糖鎖を合成することができる。このような手法を本著者らは「バイオコンビナトリアル合成法」と呼んでいる。これまでに糖鎖プライマーを作用させた細胞は50種類を超え、検出された糖鎖の種類は200種類を超えている。糖鎖プライマーLac-C12、GlcNAc-C12およびXyl-Ser-C12を用いて得られた主な糖鎖をそれぞれ図3〜5に示した。 Lac-C12は、糖脂質の生合成経路の前駆体となる糖鎖プライマーである。そのために、主要な糖脂質であるガングリオシド、グロボ系糖脂質、およびネオラクト系糖脂質でみられる様なオリゴ糖が糖鎖プライマーに伸長していた(図 3)。B16メラノーマ細胞では酸性糖としてはGM3型の生成物のみが観察されたが、細胞の種類によってはジシアロガングリオシドやトリシアロガングリオシドが観察された。この様に細胞に発現している糖脂質と対応したオリゴ糖が糖鎖プライマー法でも検出できていた。ガングリオ系列の糖鎖ではグリコイル型のシアル酸が結合した構造も観察された。
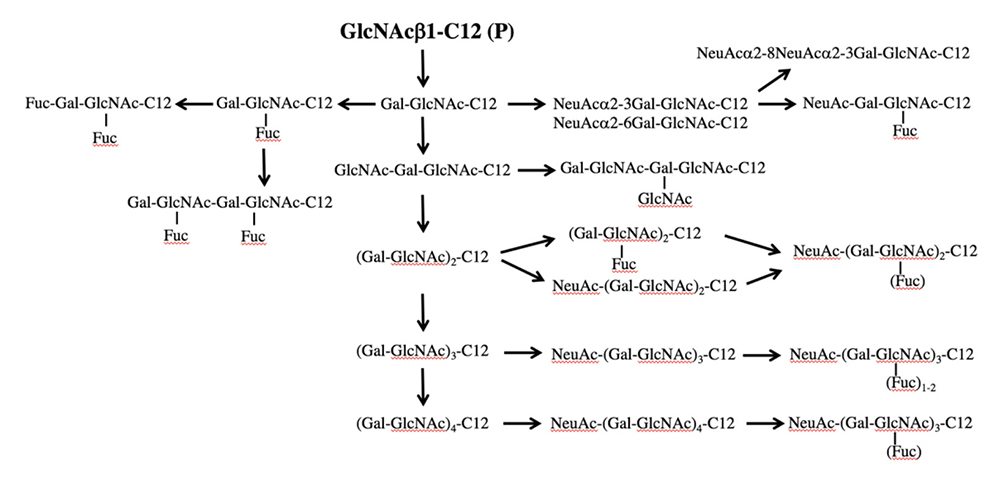
GlcNAc-C12はラクトサミンやシアル化ルイス抗原などラクト・ネオラクト系列の生合成経路の前駆体である。GlcNAc-C12をHL60細胞などと相互作用させると、ルイスX、シアル化ルイスX、ポリラクトサミンやシアリルポリラクトサミンなどが得られた(図 4)12。ラクトサミンへのシアル酸の結合様式はα2-3とα2-6の両方が含まれており、LC-MSで区別して検出された。ネオラクト系列の糖鎖を得るための糖鎖プライマーとしてGalβ1-4GlcNAc-C12(LacNAc-C12)も用いた。その結果、LacNAc-C12でもGlcNAc-C12と同様の生成物が得られていたが、収量は半分程度に低下していた。糖鎖プライマーの親疎水バランスの影響があると思われるので改良の余地はあるが、現在のところ、ネオラクト系列の糖鎖を得るための糖鎖プライマーは単糖型のGlcNAc-C12が適していると考えている。
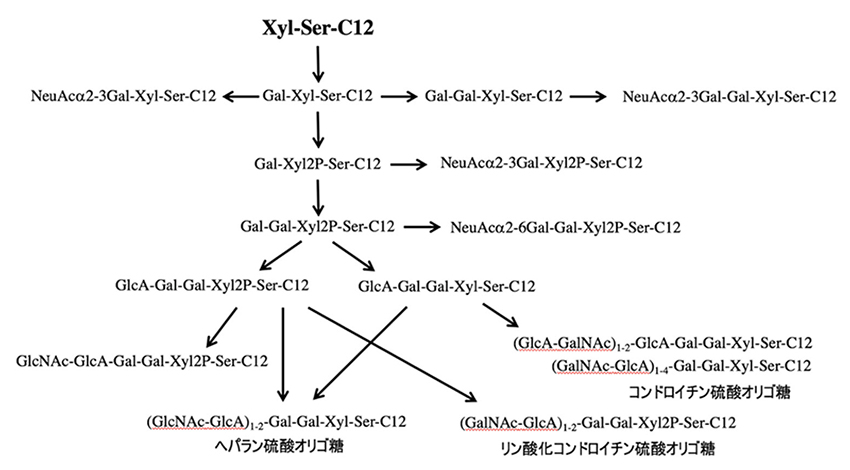
糖鎖プライマーXyl-Ser-C12はプロテオグリカン型糖鎖の生合成の前駆体である。プロテオグリカンの生合成では、Xyl残基にGal-Galが付加され、その先にHexAやHexNAcが繰り返して伸長され、(HexA-HexNAc)n-Gal-Gal-Xyl構造が形成される。CHO細胞にXyl-Ser-C12を相互作用させると、Gal-Gal-Xyl-Ser-C12が生成され、次にHexNAcとHexAの伸長した(HexNAc-HexA)n-Gal-Gal-Xyl-Ser-C12が検出されたことから、プロテオグリカン型の糖鎖が得られたことが示された(図 5)。CHO細胞では、最長でn=4で糖鎖プライマーに10糖の伸長がみられ、生成物の種類は14種類であった13。Xyl-Serがプロテオグリカン型糖鎖の生合成で有利であることを示すために、Xyl-pNPとの比較実験を行った。CHO細胞にXyl-pNPを相互作用させたところ、Xyl-Ser-C12と同じ条件で検出された糖鎖伸長生成物は5種類のみであった。Xyl-Ser-C12において Ser残基が存在することは、糖鎖合成という観点では有用であった。そこで、SerをThrに変えたXyl-Thr-C12を合成したところ、糖鎖伸長が殆ど見られず、キシロースの結合するアミノ酸残基がSerであることは、糖転移酵素の認識に重要であることが示唆された15。さらに、Xylの結合するアミノ酸のコンセンサスモチーフを考慮して、Gly-Serをアミノ酸残基に有するGly-(Xyl)Ser-C12を設計した。その結果、Xyl-Ser-C12ではNeuAc-Gal-Xyl-Ser-C12の様なシアリルオリゴ糖が検出されていたが、Gly-(Xyl)Ser-C12ではシアリル化反応が抑制されていた15。NeuAcの修飾によりプロテオグリカン型糖鎖の伸長が阻害されると考えられているが、Gly-Serによりシアル酸の転移が抑制されることでプロテオグリカン型糖鎖の伸長が優先的に起きることが示された。
正常ヒト皮膚繊維芽NHDF細胞では、シアリルオリゴ糖としてはNeuAcα2-3Gal-Xyl-Ser-C12が主要生成物であり次にNeuAcα2-3Gal-Xyl2P-Ser-C12が検出されていた15。これに加えてGAG鎖合成の中間体であると推察されるNeuAcα2-6Gal-Gal-Xyl-Ser-C12という構造も検出されていた。一方、GAG鎖の合成経路では、リン酸化されたHexNAc-HexA-Hex-Hex-Xyl2P-Ser-C12が最も多く検出された。また、HexNAc-HexA が繰り返された(HexNAc-HexA)2-Hex-Hex-Xyl2P-Ser-C12が生成れており、この生成物はC-ABCで分解されるが、ヘパリチナーゼでは分解されないことから、コンドロイチン硫酸型(GalNAc-GlcA)n-Gal-Gal-Xyl2P-Ser-C12のGAG鎖が伸長していることが示された。一方、Xylが脱リン酸化された(HexNAc-HexA)2-Hex-Hex-Xyl-Ser-C12では、C-ABCとヘパリチナーゼの両方から加水分解されていた。これにより、脱リン酸化されたコンドロイチン硫酸型(GalNAc-GlcA)n-Gal-Gal-Xyl-Ser-C12やヘパラン硫酸型(GlcNAc-GlcA)n-Gal-Gal-Xyl-Ser-C12のGAG鎖が伸長されていると推察された。LC-MSのピーク強度からはコンドロイチン硫酸型が主要な産物であった。これによりXylがリン酸化されているとコンドロイチン硫酸が伸長するが、ヘパラン硫酸型が伸長するにはXylの脱リン酸化が必要であることと示唆された。これにより、このように、GAG鎖の伸長反応におけるキシロースのリン酸化の役割が明らかになり、GAG鎖の生合成経路の中間体を検出することで、生合成のメカニズム解析に有効であると考えられる。
さらに、GalNAcを有する糖鎖プライマーとしてGalNAc-Thr-C12あるいはGalNAc-Ser-C12をムチン型糖鎖の獲得を目的として合成した。複数の細胞と相互作用させたところ、シアリルTn抗原、T抗原、シアリルT抗原、フコシルT抗原を始めとして、多くの細胞で主にコア1やコア2に分類される生成物が検出されている。これらの糖鎖プライマーはムチン型の糖鎖ライブラリーの構築に活用できると期待している。
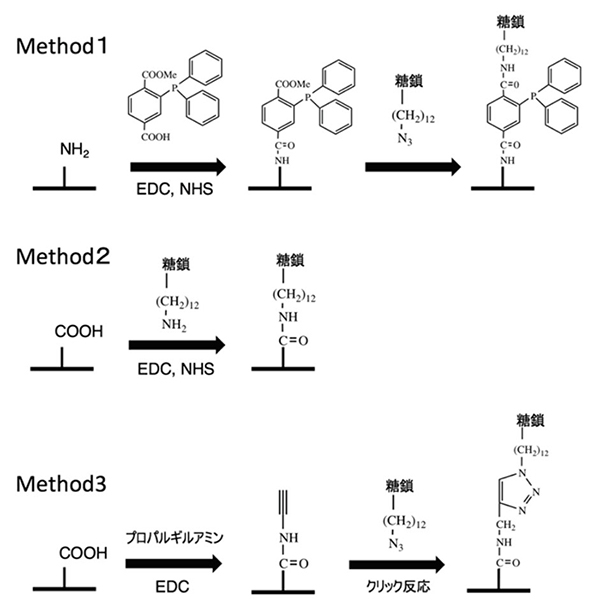
糖鎖プライマー法により多様な糖鎖を合成できることから、糖鎖ライブラリーの構築が可能になってきた。得られた糖鎖の認識を評価するにはセンサーなどに固定化する必要がある。そのためにアジド化された糖鎖プライマーを開発した18。アグリコン末端に導入されたアジド基は糖鎖伸長反応に影響しない。これにより細胞はアジド基を有した糖鎖ライブラリーを合成してくれる。アジド化糖鎖ライブラリーは、化学的に固相表面に固定化できる。その方法としては、図 6に示す様な、1)Schtaudinger反応19、2)アジド基をアミノ基に還元したのちに縮合反応19、および3)クリック反応を行なっている。
エイズウイルス(HIV)の感染では、エンベロープタンパク質(gp120やgp160)と糖脂質の結合によりHIV と生体膜との融合が誘起されることが知られている20。特に、gp120はGM3、GalCer、あるいはGb3Cerと親和性を有することが報告されている21-23。そこで、Lac-C12-N3をマウスB16メラノーマ細胞と相互作用することで、Gb3を有するGal-Gal-Glc-C12-N3やGM3型のNeuAc-Gal-Glc-C12-N3を合成した。この化合物のアジド基をアミノ基に還元することで、センサー基板のカルボキシ基と縮合反応で固定化した。固定化した糖鎖とgp120やgp160との相互作用を表面プラズモン共鳴装置を用いて定量的に評価した。その結果、gp160はGM3型よりもGb3やLacに親和性を示し、gp120ではどの糖鎖に対しても親和性は低く同程度であることが示された24。
最近では、糖鎖プライマー法により得られた糖鎖ライブラリーをクリック反応で微粒子表面に固定化することで、インフルエンザウイルスを検出することにも成功している。このように、糖鎖プライマー法ではアジド化された糖鎖ライブラリーを簡便に合成することが可能であり、それを活用した糖鎖認識の評価に活用できる。
以上で述べた様に糖鎖ライブラリーの構築を目指して糖鎖プライマー法の研究を進めてきた。一方で、糖鎖プライマー法では細胞の糖鎖生合成経路に従った糖鎖伸長生成物を少ない細胞数で検出できることから、細胞機能と関係した糖鎖の発現解析に適用することも可能である。これまでのいくつかの実施例を以下に紹介する。
神経芽腫は5歳以下の小児での発生率が高い神経細胞に発生するがんである。神経芽腫細胞では正常の神経細胞と比較してGD2を高発現している25。また、ガングリオシドの発現と予後との関係が知られており、ガングリオb系列のガングリオシドが発現している場合には予後は良好であり26、一方、ガングリオb系列のガングリオシドの発現が低い場合には予後不良であるということが報告されている27。
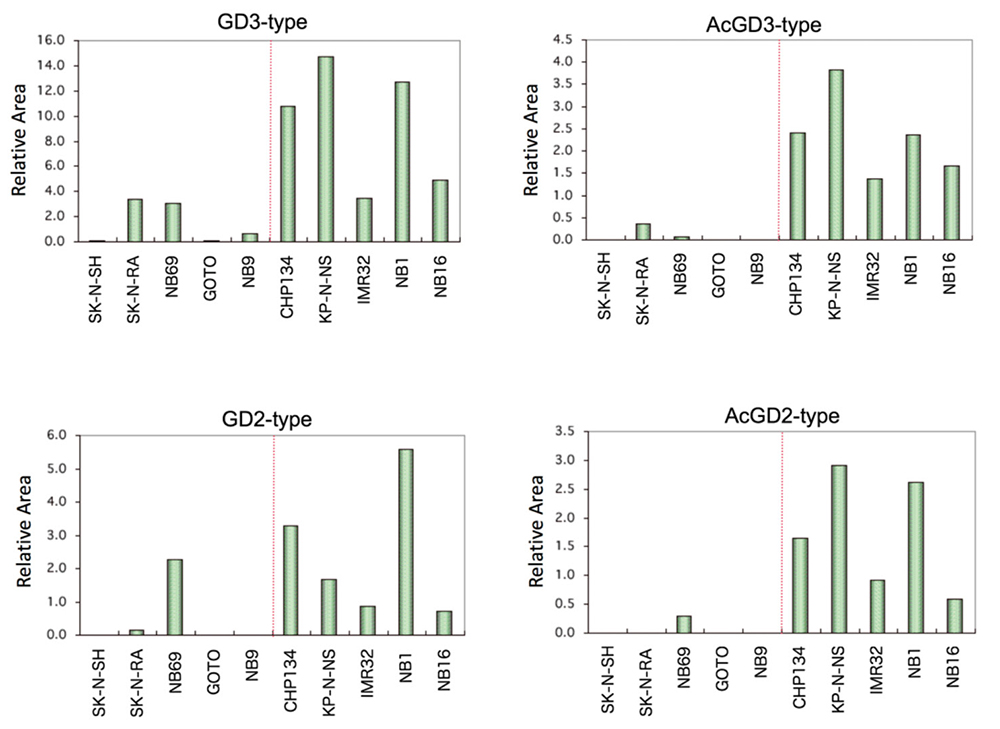
我々は、糖鎖プライマーLac-C12を神経分化マーカー(Phox2a/b、TrkC、NCAM、Neurofilamentなど)の発現量の異なる神経芽腫細胞株10種類と相互作用させて、糖鎖伸長生成物の構造解析を行なった。糖鎖伸長生成物としてガングリオ系列やグロボ系列など16種類が検出された。その中でGD3型やGD2型とそのアセチル化体の生成物の発現量が高い細胞株では、神経分化マーカーの発現が高かった(図 7)。同じ細胞株を用いて内在性糖脂質の解析を行ったところ、GD2とアセチル化GD2で神経分化マーカーの発現との対応がみられていた28。また、糖鎖合成遺伝子の解析を行ったところ、神経分化マーカーの発現が高かった細胞株ではGD3合成遺伝子ST8SIA1の発現が高かった。一方で、GalNAc転移遺伝子B4GALNT1、Gal転移遺伝子B3GALT4およびシアル酸転移遺伝子ST3GAL1/4の発現は神経分化マーカーとの関連は見られなかった。これにより、神経芽腫におけるガングリオb系列の糖鎖の発現の向上はST8SIA1によりGD3の発現の向上が影響しているものと考えられた。
ヒト急性骨髄性白血病細胞HL60細胞は単球様あるいは顆粒球様細胞への分化能を有している。
例えば、12-O-acetyldecanoylphorbol-13-acetate(TPA)で処理することで単球様細胞に分化する。これによりGM3が顕著に増加し、ネオラクト系糖脂質nLc4が減少することが知られている29。そこで、Lac-C12とGlcNAc-C12を分化誘導前後のHL60細胞と相互作用させて、得られる糖鎖伸長生成物の変化をLC-MSで測定した。細胞はTPA添加後12時間で接着する様になり、72時間で非特異的エステラーゼ活性が観察されたことから分化誘導が確認された。細胞の接着挙動が見られた頃に、Lac-C12由来の生成物ではGM3型(NeuAc-Gal-Glc-C12)とGb3(Gal-Gal-Glc-C12)の検出量が向上していた。一方、GlcNAc-C12由来の生成物では、Gal-(Fuc)GlcNAc-C12の合成量が減少し始めた。この様な糖鎖の発現量の変化は、GM3合成遺伝子、Gb3合成遺伝子、FUT4などの遺伝子の発現量の変化と相関していた。TPA添加後に、GM3型の発現がまず上昇し、遅れてGb3の発現が上昇し、非特異的エステラーゼ活性が確認できた時点では両者の発現量は飽和していた。GM3型とGb3が増加するにつれてGal-(Fuc)GlcNAc-C12の減少が始まっていた。このように、糖鎖プライマー法により分化誘導試薬を加えた後の糖鎖生合成の変化を簡便にモニタリングできることが示された。
胚性癌腫細胞(Embryonic Carcinoma Cell, EC細胞)は、精巣や卵巣に自然発生する腫瘍であるテラトカルシノーマにおいて未分化性を保持する細胞である。EC細胞は初期胚の細胞と類似しているので、初期発生過程の研究における分化モデル細胞として利用されてきた。マウスEC細胞のひとつであるF9細胞はRAで処理すると近位内胚葉様の特性を示すことが知られており29、レチノイン酸(RA)とdibutyryl cyclic AMP(dcAMP)で処理すると遠位内胚葉様の特性を示す30。
F9ではSSEA1/3やForssman抗原が発現していることが知られている。F9をRAで分化誘導するとSSEA131とForssman抗原32が減少し、SSEA333やガングリオシド29,34は増加することが知られている。我々は、F9細胞にRAとdcAMPを作用させて分化誘導したF9細胞にGlcNAc-C12を相互作用させることで糖鎖生合成経路の変化を観察した35。その結果、GlcNAc-C12より得られるネオラクト系列の糖鎖の合成量が全体的に増加していた。特に、NeuNAc-Gal-GlcNAc-C12が顕著に増加していた。さらに、得られた糖鎖の合成酵素の遺伝子として、Galβ1-4GlcNAcを合成するB4galt1やNeuAcα2-3Galを合成するSt3gal6の発現の向上も見られていた。同時に、内在性糖脂質を抽出して質量分析装置で解析したところ、パラグロボシド(nLc4Cer)やシアリルパラグロボシドの発現が向上していることが示された。
がん細胞の悪性化の要因としての転移性と糖鎖との関係はよく研究されている36-41。例えば、がん細胞と血管内皮細胞との相互作用にはシアリルLex抗原や硫酸化シアリルLex抗原が、組織内での浸潤にはガングリオシドや硫酸化糖脂質が関与している。また、ヘパラン硫酸もがん細胞の増殖や転移性に関与することが知られている。我々は、マウス骨肉腫細胞の転移性に関わる糖鎖の解明を行った。そのために、高転移性のマウス骨肉腫細胞FBJ-LLと低転移性のFBJ-S1細胞にXyl-Ser-C12を相互作用させて糖鎖解析を実施した15。これら骨肉腫細胞からはGal-Gal-Xyl-Ser-C12にHexAとHexNAcが繰り返して結合した糖鎖伸長生成物が得られていた。このような生成物はヘパリチナーゼで加水分解されたことから、生成物はヘパラン硫酸型のGAGであると推察された。FBJ-LL細胞とFBJ-S1細胞ではほぼ同一の生成物が得られたが、高転移性のFBJ-LL細胞ではGAG型の生成物の量が有意に低かった。GAG鎖を伸長する酵素(Ext1やExt2)の発現をRT-PCRやウエスタンブロッティングで評価したところ、FBJ-LL細胞ではFBJ-S1細胞と比較して低いことが示され、糖鎖伸長生成物の検出量との対応が見られた。そこで、Ext1の発現が高かったFBJ-S1細胞でExt1遺伝子をsiRNA法でノックダウンしたところ、細胞の遊走性が向上した。さらに、予期せぬことにExt1遺伝子をノックダウンしたFBJ-S1細胞では、ヘパラナーゼの発現の増加も観察された。この様に、ヘパラン硫酸の発現には合成と分解が連動していることが示された。このように、糖鎖プライマー法により細胞の遊走性と糖鎖との関係を検討することができた。
本著者は糖鎖プライマー法の活用法として、大きく分けて二つの方法を検討してきた。一つは糖鎖ライブラリーの作成であり、もう一つは細胞で発現する糖鎖のプロファイリングである。両者は、糖鎖プライマーを細胞と相互作用させて、糖鎖伸長生成物をLC-MSで解析するという実験を行うことで、同時に達成することができる。糖鎖ライブラリーが得られることで、たんぱく質やウイルスに対する糖鎖認識の解析およびLC-MSでの糖鎖構造解析のためのデータベースの作成などに活用している。一方、多様な細胞での糖鎖プロファイリングを行うことで細胞に発現する糖鎖をデータベース化しており、さらにバイオインフォマティックスの手法により細胞間での比較解析および細胞機能との相関解析へと展開している。これにより、細胞の種類や機能に依存した糖鎖を特定することができ、またそのような糖鎖の合成に関わる遺伝子の解析に効率よくアプローチできるようになってきた。本著者の提案する糖鎖プライマー法がグライコミクスの発展に寄与できる新たなツールとなることを期待している。



