Toda T, Kobayashi K, Takeda S, Sasaki J, Kurahashi H, Kano H, Tachikawa M, Wang F, Nagai Y, Taniguchi K, Taniguchi M, Sunada Y, Terashima T, Endo T, Matsumura K (2003) Fukuyama-type congenital muscular dystrophy (FCMD) and alpha-dystroglycanopathy. Congenit Anom (Kyoto). 43, 97-104.
Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T (1997) Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J Biol Chem. 272, 2156-2162.
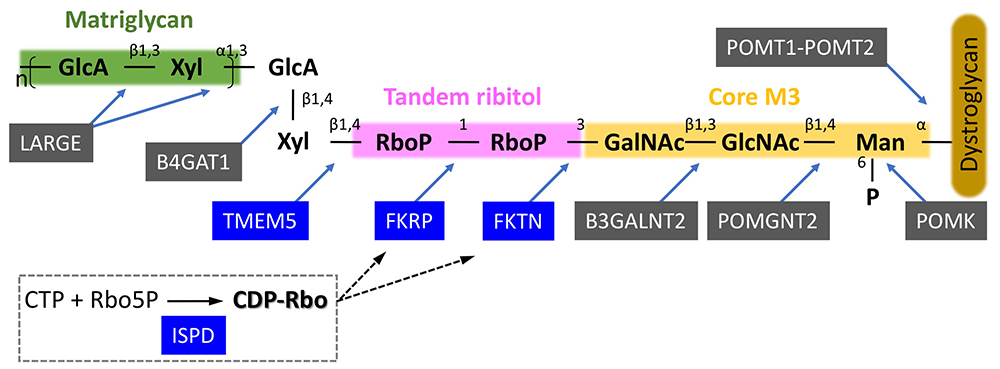
Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, Campbell KP (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 335, 93-96.
Yoshida-Moriguchi T, Willer T, Anderson ME, Venzke D, Whyte T, Muntoni F, Lee H, Nelson SF, Yu L, Campbell KP (2013) SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science. 341, 896-899.
Kanagawa M, Kobayashi K, Tajiri M, Manya H, Kuga A, Yamaguchi Y, Akasaka-Manya K, Furukawa JI, Mizuno M, Kawakami H, Shinohara Y, Wada Y, Endo T, Toda T (2016) Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 14, 2209-2223.
Manya H, Yamaguchi Y, Kanagawa M, Kobayashi K, Tajiri M, Akasaka-Manya K, Kawakami H, Mizuno M, Wada Y, Toda T, Endo T (2016) The muscular dystrophy gene TMEM5 encodes a ribitol β1,4-xylosyltransferase required for the functional glycosylation of dystroglycan. J Biol Chem. 291, 24618-24627.
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 394, 388-392.
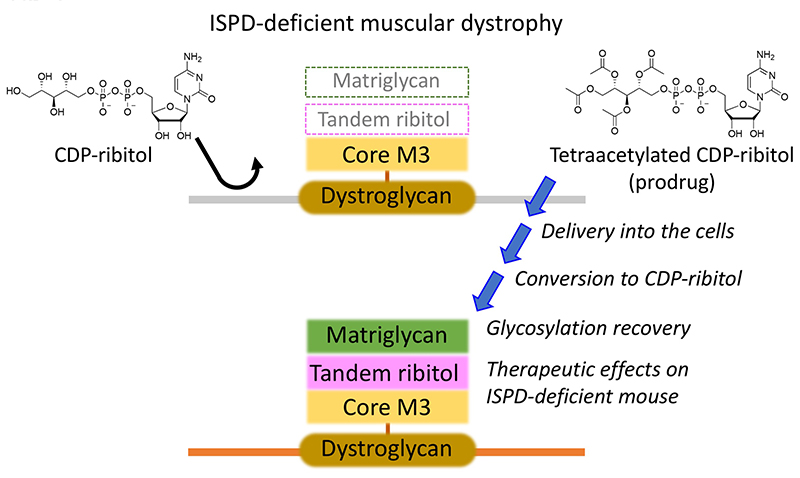
Gerin I, Ury B, Breloy I, Bouchet-Seraphin C, Bolsée J, Halbout M, Graff J, Vertommen D, Muccioli GG, Seta N, Cuisset JM, Dabaj I, Quijano-Roy S, Grahn A, Van Schaftingen E, Bommer GT (2016) ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat Commun. 7, 11534.
Cataldi MP, Lu P, Blaeser A, Lu QL (2018) Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun. 9, 3448.
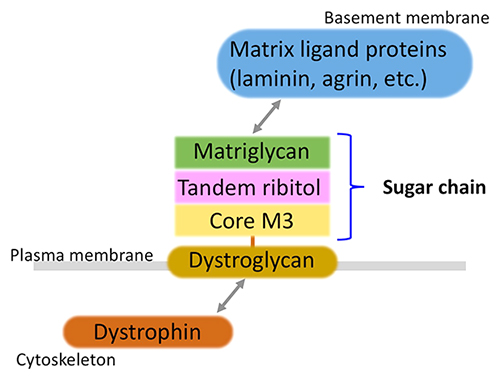
Kanagawa M (2021) Dystroglycanopathy: From elucidation of molecular and pathological mechanisms to development of treatment methods. Int J Mol Sci. 22, 13162.