
Sheng-Tao Li
Research Scientist at Glycometabolic Biochemistry Laboratory, RIKEN Pioneering Research Institute (PRI).
He received his PhD in 2018 at the University of Jiangnan in China, and then joined the RIKEN Pioneering Research Institute as a postdoctoral fellow. He became a Research Scientist at RIKEN in 2025. His research interests focus on the biosynthesis and degradation of N-glycosylation.
N-glycosylation, as the most important co-/post-translational modification of proteins, begins with the assembly of dolichol-linked oligosaccharide (DLO) on the ER membrane. Eukaryotic cells rarely accumulate DLO, however, immature and mature DLO structures are accumulated under some genetic and environmental conditions, thereby increasing a risk of abnormal N-glycosylation. Here, I describe that identify a novel gene encoding DLO pyrophosphatase responsible for the degradation of these DLO.
In eukaryotes, asparagine (N)-glycosylation starts with a multistep process sequentially assembling a dolichol-linked oligosaccharide (DLO) precursor on the endoplasmic reticulum (ER) membrane, by a series of glycosyltransferases encoded by ALG (asparagine linked glycosylation) genes1. Once assembled, the Glc3Man9GlcNAc2 oligosaccharide is transferred to nascent polypeptides by oligosaccharyltransferase (OST) through an N-glycosidic bond to an asparagine residue of the Asn-X-Ser/Thr (where X represents any amino acid, except for Pro) consensus sequence2,3.
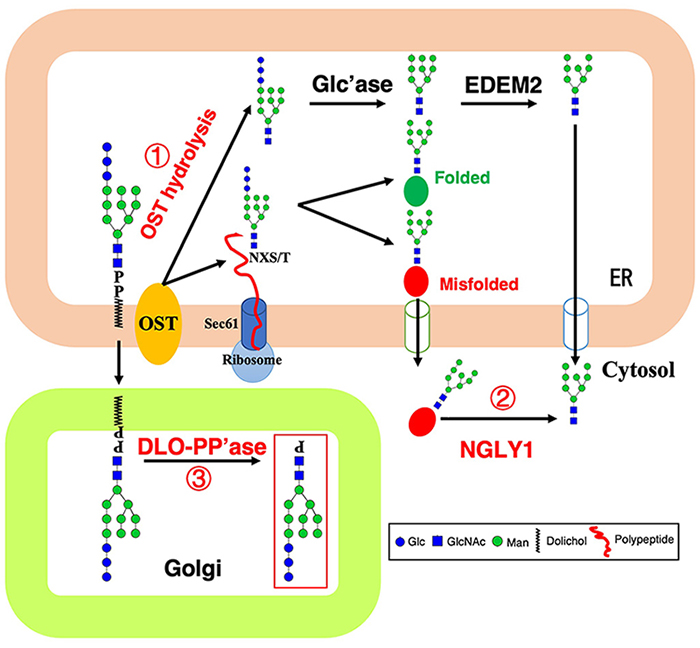
Since the 1970s-1980s, studies have shown that the synthesis of N-glycosylation is accompanied by degradation into free oligosaccharides, which are divided into neutral oligosaccharides (NOS) and phosphorylated oligosaccharides (POS)4. The following decades gradually clarified that NOS originated from the hydrolytic activity of OST on DLO (Figure 1, reaction 1) and the removal of oligosaccharides from misfolded glycoproteins by Ngly1 (Figure 1, reaction 2), and POS originated from DLO by the action of a pyrophosphatase (DLO-PP’ase) (Figure 1, reaction 3)5. DLO-PP’ase activity has been detected in both yeast and mammals6-13, and its activity is mainly present in the Golgi of mammalian cells14,15. However, the gene encoding this enzyme remains a mystery in any organism.
Recent studies demonstrated that POS mainly originated from immature DLO intermediates that are accumulated when DLO biosynthesis is impaired, such as congenital disorders of glycosylation type I (CDG-I) patient-derived cells16,17, glucose-starved or 2-deoxyglucose-treated mammalian cells18,19. These results indicated that production of POS will be a highly regulated process tightly linked to DLO biosynthesis, while the lack of gene information for DLO-PP’ase keep us from clarifying its physiological function, as well as the details of regulatory mechanisms. Recently, our study identified the yeast DLO-PP’ase, Llp1, and proposed a new DLO homeostasis/quality control mechanism whereby DLO accumulated in the ER is translocated to the Golgi and degraded by Llp120.
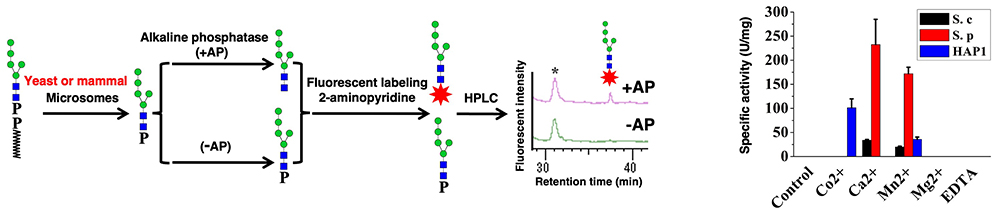
To identify the DLO-PP’ase, we developed an in vitro assay to detect DLO-PP’ase activity as follows: POS (Man5GlcNAc2-P) was produced by incubating the DLO (Man5GlcNAc2-PP-Dol) with the detergent-solubilized yeast or mammalian microsomes, which contain DLO-PP’ase activity. Man5GlcNAc2-P was purified by anion exchange, and fluorescently (2-aminopyridine) labeled after treatment with alkaline phosphatase, then analyzed by HPLC (Figure 2). We compared the DLO-PP’ase activities of yeast (S. cerevisiae and S. pombe), and human cell (HAP1) microsomes and found that yeast and human enzymes had distinct patterns with regard to the effect of divalent cations (Figure 2). This finding suggests that yeast and mammalian DLO-PP’ase may be encoded by distinct genes.
To identify a gene encoding yeast DLO-PP’ase, yeast microsomes were used as the material for purifying the enzyme. Based on the partial purification of enzyme, followed by shotgun analysis of purified protein fractions, candidates were selected and screening from yeast knockout strains library. Among these candidate gene mutants, one of the mutants, YJR112W-AΔ, exhibited neither POS in cells nor DLO-PP’ase activity. The YJR112W-A gene was named LLP1 after lipid-linked oligosaccharide pyrophosphatase.
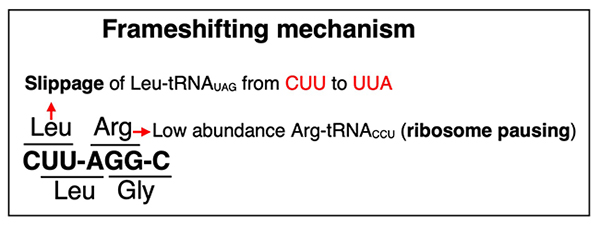
LLP1 is registered in the Saccharomyces Genome Database (SGD), and it is annotated as the gene with an intronic region (as of January 2026). However, we found that the intron-deleted protein was inactive, suggesting that the annotation of the intronic region is inaccurate. Using ribosome profiling data, we found that LLP1 use a programmed +1 frameshift similar to yeast retrotransposon gene which occurs in the CUUAGGC (C64–C70 in LLP1) motif through a translational pause caused at 0-frame AGG — a rare codon — by the low availability of tRNAArgCCU21,22, followed by a +1-slip of tRNALeuUAG from CUU to UUA on the P site of ribosome (Figure 3)23,24. These results indicated that programmed +1 translational frameshifting in LLP1 occurs through a tRNA slippage.
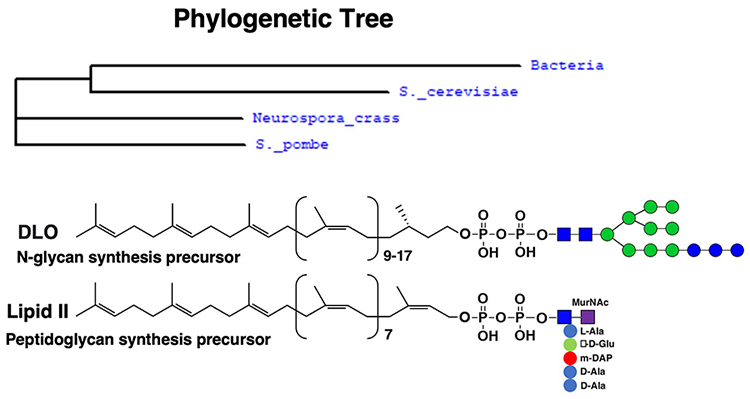
Analysis of Llp1 protein sequence showed that Llp1 belongs to the VanZ-like family proteins. We investigated the phylogenetic distribution of the VanZ family and discovered that it is broadly conserved in Gram-positive bacteria and various fungi (Figure 4), but not found in plants or vertebrates in our analyses. The VanZ gene was first identified in clinically isolated Enterococcus faecium BM4147, which is resistant to glycopeptide antibiotic vancomycin25-27. Based on the similarity between DLO and bacterial lipid II structures (Figure 4), it seemed reasonable that VanZ is likely involved in the cleavage of lipid II.
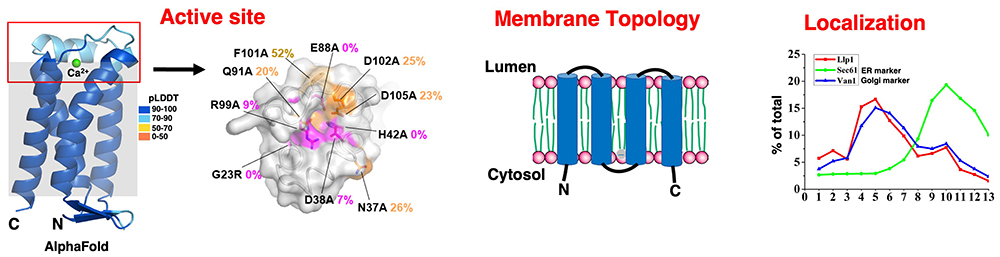
Through the homology alignment of VanZ family proteins among different species and alanine scanning mutagenesis of the conserved residues, we identified several conserved domains. The three-dimensional (3D) model of Llp1 (S. cerevisiae) predicted by AlphaFold version 3.028 showed that these domains are located on one side of the membrane and are rich in positively and negatively charged residues, which may constitute a phosphatase active site (Figure 5). Using the TEV protease protection assay, we demonstrated that the N and C termini of the Llp1 protein face the cytosol and the active site of Llp1 is located at the luminal side (Figure 5). Through sucrose density gradient fractionation, the distribution of Llp1 was similar to the Golgi marker Van1 but differed from the ER marker Sec61 (Figure 5). These results indicated that Llp1 is mainly localized at the Golgi fraction, which is consistent with the localization of mammalian DLO-PP’ase14.
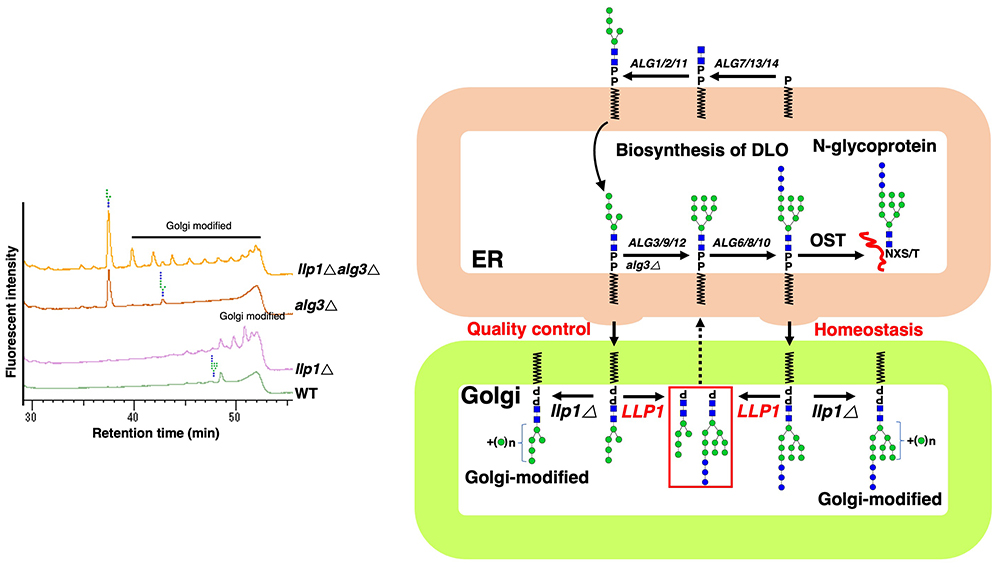
DLO biosynthesis occurs in the ER, whereas Llp1 is localized in the Golgi, implying that DLO in the ER needs to be translocated to the Golgi. Although the presence of DLO in the Golgi has not been reported, biochemical studies found that in rat liver, the content of dolichol/dolichol fatty acyl esters in Golgi was ~10 times that in the ER, and a large amount of dolichol-P was also found in Golgi29,30. We hypothesized that, under normal conditions, DLO in the Golgi could be rapidly degraded by Llp1. To test this, the DLO structure in the llp1Δ and llp1Δ alg3Δ strain was examined. Unexpectedly, Golgi mannosyltransferase-modified DLO structures were found in llp1Δ and llp1Δ alg3Δ cells (Figure 6). These results indicated that, although DLO biosynthesis occurs in the ER, surplus/incompletely assembled DLO is translocated to the Golgi to be eliminated by Llp1, thus playing a role in DLO homeostasis/quality control (Figure 6).
In summary, our study identified the yeast DLO-PP’ase, Llp1, and proposed a new DLO homeostasis/quality control mechanism whereby DLO accumulated in the ER is translocated to the Golgi and degraded by Llp1. Future studies on the precise molecular mechanisms of how DLO accumulated in the ER are recognized and translocated to the Golgi, and how Dolichol-P produced in the Golgi returns to the ER will be of great significance. On the other hand, there is no homologous Llp1 protein in mammals, suggesting that mammals may have different POSs production systems, and different DLO-PP’ase should be involved. Identification of the DLO-PP’ase gene in mammals is urgently needed in the future.



