この作品はクリエイティブ・コモンズ 表示 4.0 |
 |
|---|
糖鎖異常型筋ジストロフィー(ジストログリカン異常症)の病態と治療法 | ||||||||||||||
 |
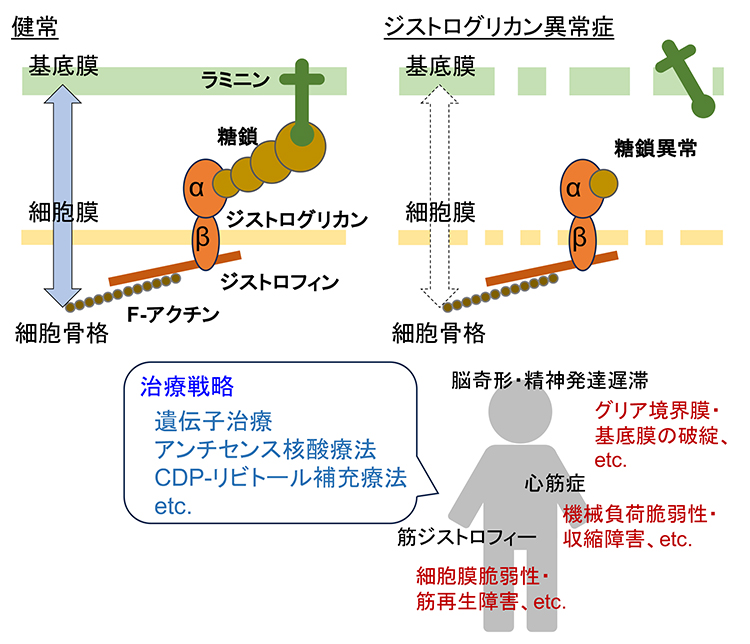
筋ジストロフィーは筋線維の壊死・再生を主病変とし、進行性の筋力低下を認める遺伝性疾患の総称で、非常に多くの原因遺伝子が存在し病型も多岐にわたる。糖鎖異常型筋ジストロフィー(ジストログリカン異常症:DG異常症)は、基底膜ラミニン受容体であるジストログリカン(DG)の糖鎖異常を発症要因とする疾患群である。日本におけるDG異常症では、フクチン(FKTN)遺伝子を原因とする福山型筋ジストロフィー(FCMD)が大半を占める。 DGはα鎖とβ鎖からなる。α-DGは細胞外でラミニンと結合する。膜貫通型のβ-DGはα-DGを細胞表面に繋ぎとめておく一方で、細胞内ではアクチン結合分子であるジストロフィンと結合する。α-DGとラミニンとの結合には糖鎖修飾が必要であることから、糖鎖異常によって、ラミニン(基底膜)-DG-ジストロフィン-アクチン(細胞骨格)の連携が破綻し、筋線維が脆弱化、筋ジストロフィーが発症する(図 1)。発症に関与する糖鎖構造と生合成酵素の詳細は、“哺乳類におけるO-マンノース型糖鎖の構造と生合成機構”の項を参照されたいが、ラミニン結合能をもつ糖鎖は、キシロースとグルクロン酸の二糖の繰り返し構造で、現在はマトリグリカンと呼ばれている。また、リビトールリン酸という糖アルコールリン酸が二つ連なったユニークな構造も存在し、マトリグリカンが伸長するために必要な土台構造となっている。フクチンはリビトールリン酸転移酵素であり、FCMD患者ではリビトールリン酸構造が形成されないため、マトリグリカン修飾も進行せず発症に至る。
DG異常症患者では、筋病変に加えて、精神発達遅滞や脳奇形(Ⅱ型滑脳症)などの中枢神経障害や心筋症を伴うことがある。筋線維は損傷すると、衛星細胞とよばれる幹細胞が活性化し、筋前駆細胞、筋芽細胞へと分化、そして筋管へと融合することで筋線維は再生する。筋線維選択的なmuscle creatine kinase (MCK)-フクチン-コンディショナルノックアウト(cKO)マウスは、機械的負荷に対して筋細胞膜が脆弱化しているものの、筋病理学的にはマイルドな所見にとどまる。一方、筋前駆細胞選択的なMyf5-フクチンcKOマウスは、重篤な筋ジストロフィー所見を示す。Myf5-フクチンcKOマウスから調製した衛星細胞は増殖能・分化能とも低下しており、その結果、筋再生能も低下していた(1)。つまり、筋再生力の低下がDG異常症の重篤な筋病変の原因のひとつと考えられる。脳奇形については、胎児期の脳形成過程における脳表グリア境界膜-基底膜複合体の維持にDG糖鎖が重要とされており、糖鎖異常によってグリア境界膜-基底膜複合体が破綻し、神経細胞が迷出することが原因のひとつと考えられている(2)。DGが抑制性シナプス機能、軸索ガイダンス、グリア細胞エンドフィートの構造・機能に関わることも示唆されており、これらの障害が病態に関与していることも考えられる。心筋においては、筋細胞膜の物理的強度の維持に関わるとされる一方、血行動態負荷に対する適応的肥大応答に関わることも報告されている(3)。 DG異常症に分類される各疾患(例えばFCMD)は単一遺伝子疾患であるため遺伝子治療が有効と考えられる。筋再生に異常があるMyf5-フクチンcKOマウスに対して、アデノ随伴ウイルスベクターとMCKプロモーターを用いた筋線維を標的とするフクチン遺伝子治療によって治療効果が得られている(1)。この結果は、筋再生力が低下したとしても、発症の引き金となる筋線維細胞膜の脆弱性を抑制することで治療につながることを示唆している。多くのFCMD患者では、フクチンの3’非翻訳領域へのトランスポゾン挿入変異が認められる。トランスポゾン挿入によってスプライシング異常が生じ、正常なフクチンタンパク質が生成されない。スプライシング異常を是正するようなアンチセンス核酸を投与することで、モデルマウスや患者細胞で正常フクチンタンパク質が産出されるようになり、治療に繋がる可能性が報告されている(4)。 リビトールリン酸転移酵素はドナー基質としてシチジン二リン酸 (CDP)-リビトールを用いる。CDP-リビトールはisoprenoid synthase domain containing (ISPD)、別名をCRPPA (CDP-L-ribitol pyrophosphorylase A)という酵素によって合成される。ISPD変異型DG異常症は、CDP-リビトールの生合成障害が発症要因であるため、CDP-リビトールを補充することが治療につながると考えられる。最近、筋組織への送達性を高めたプロドラッグ化CDP-リビトールの投与によってISPD欠損マウスの筋病変が改善されるとの報告がなされた(5)。以上紹介したように、この10年ほどの間にDG異常症の原因遺伝子機能や分子病態機序に基づく様々な治療戦略が提唱されてきている。その発見から長年にわたり治療法がない難病とされてきたDG異常症に有効な治療法が確立される日も遠くないと期待される。 金川 基(愛媛大学大学院医学系研究科)
2024年 3月15日   | |||||||||||||