
Satomi Nadanaka
Associate professor, Laboratory of Biochemistry, Kobe Pharmaceutical University (Laboratory of Prof. Hiroshi Kitagawa)
1999: PhD, Department of Biochemistry, Kobe Pharmaceutical University (Laboratory of Prof. Kazuyuki Sugahara)
1999 to 2001: Researcher at Nippi Research Institute of Biomatrix
2001-2007: Postdoctoral Researcher in Kazutoshi Mori’s group of Kyoto University
2007: Assistant professor, Department of Biochemistry, Kobe Pharmaceutical University
2018: Associate professor, Laboratory of Biochemistry, Kobe Pharmaceutical University
Dr. Nadanaka seeks to understand how sugar signaling controls the cell function and how the rewiring of the sugar signaling pathway leads to, or prevents the progression of disease.

Hiroshi Kitagawa
Professor, Laboratory of Biochemistry, Kobe Pharmaceutical University
1991: PhD, Faculty of Pharmaceutical Sciences, Kyoto University (Laboratories of Prof. Ikuo Yamashina and Prof. Toshisuke Kawasaki)
1991 to 1994: Postdoctoral Researcher at Cytel Corporation and Scripps Research Institute (Dr. Paulson’s laboratory).
1994: Assistant professor, Department of Biochemistry, Kobe Pharmaceutical University
2000 Associate professor, Department of Biochemistry, Kobe Pharmaceutical University
2005 Full professor, Department of Biochemistry, Kobe Pharmaceutical University
2014 Board member, Kobe Pharmaceutical University
2018 Vice-president, Kobe Pharmaceutical University
Awards:
1999: The young scientist award of the Japanese Society of Carbohydrate Research
2001: The PSJ (Pharmaceutical Society of Japan) award for young scientists
2002: The young investigator award of the Japanese Biochemical Society
2013: The PSJ award for Divisional Scientific Promotions
Dr. Kitagawa works on the functions and the control of the biosynthesis and degradation of sulfated glycosaminoglycans to clarify the causes of various disorders.
Chondroitin sulfate (CS) chains are distributed on the surfaces of virtually all cells and throughout most extracellular matrices; they are covalently attached to serine residues of core proteoglycan proteins. CS chains consist of N-acetylgalactosamine (GalNAc) residues alternating in glycosidic linkages with glucuronic acid residues. During biosynthesis of CS, GalNAc residues are sulfated to varying degrees at 4- and/or 6-positions. Recent studies indicate that CS chains encode important functional information via introduction of position-specific sulfate groups. These specific sulfation patterns are recognized by numerous proteins, including growth factors, morphogens, and adhesion molecules, and these interactions regulate key events in development, normal physiology, and pathophysiology. Here, we describe the significance of CS chains functioning as a signal molecule or a co-receptor of growth factors/morphogens in cancer biology.。
Tumor metastasis involves cancer cell invasion across basement membranes and interstitial tissues. The initial invasion step consists of adherence of the tumor cell to the extracellular matrix (ECM), and this binding transduces a variety of signals from the ECM to the tumor cell. Accordingly, it is critical to establish the mechanisms by which extracellular cues influence the intracellular activities that regulate tumor cell invasion. Among the extracellular cues are chondroitin sulfates. Chondroitin sulfate (CS), a type of glycosaminoglycan, is present on the cell surface and in the extracellular matrix (ECM). There is ample evidence for a pro-tumorigenic role of CS in enhancement of cell proliferation, cell motility, and metastasis (Fig. 1) 1-4. CS in the ECM acts on tumor cells to promote tumor progression. For example, CS interacts with tumor cells via the multifunctional transmembrane protein CD44 5,6. The interaction of CS with CD44 triggers CD44 cleavage and may thereby be involved in tumor progression 7. Furthermore, it has been reported that tumor-associated glycocalyces that include CS play a key role in promoting and regulating breast cancer progression and metastasis 8. Here, we focus on how tumor-related cell signaling and function are regulated by the sulfate motifs in CS chains.
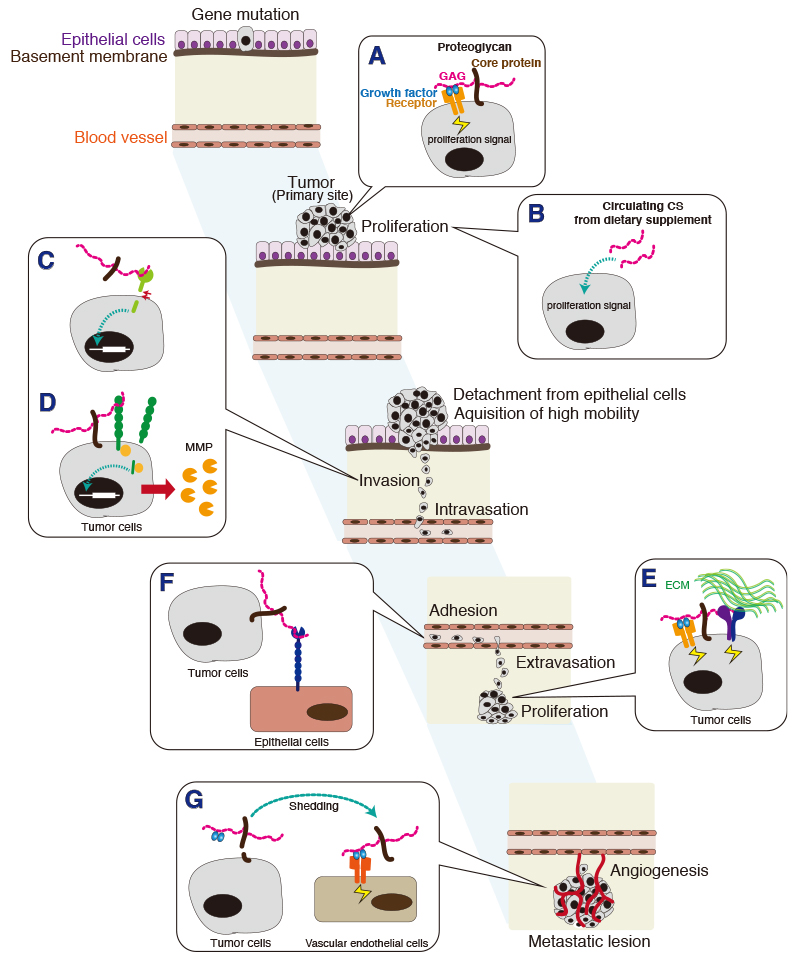
CS chains attached to a unique core protein are expressed as a proteoglycan (PG). Both core proteins and CS chains, particularly sulfation pattern of CS chains, participate in interactions with many protein ligands, such as growth factors, cytokines, and morphogens implicated in tumor progression; CS biosynthetic or degradative enzymes in a developing tumor might modify CS structure and thus alter cell function.
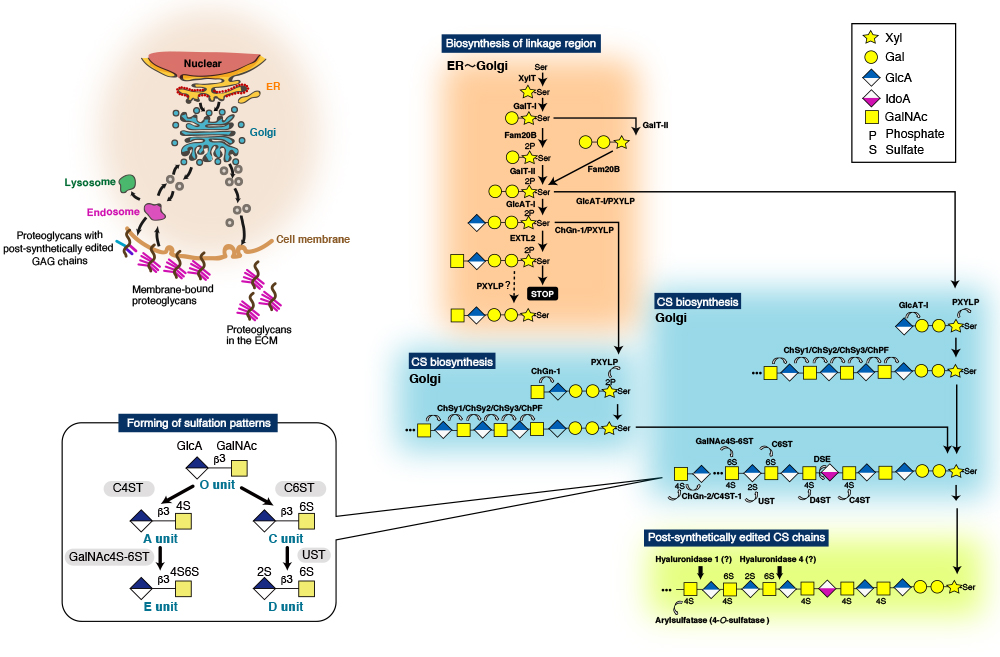
CS chains consist of repeating disaccharide units [(-4GlcAβ1-3GalNAcβ1-)n], and are covalently linked to specific serine (Ser) residues in any of the core proteins via the so-called glycosaminoglycan-protein linkage region (GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-O-Ser) (Fig. 2) 9,10. The biosynthesis of the repeating disaccharide units is constructed by a combination of six homologous glycosyltransferases - chondroitin (Chn) synthases (ChSy)-1, -2, and -3, Chn polymerizing factor (ChPF), and Chn GalNAc transferases (ChGn)-1, and -2 (Fig. 2). The resulting backbones of CS chains are subsequently modified with sulfation and uronate epimerization 9. Based on the substrate preferences of Chn sulfotransferases identified to date, the biosynthetic scheme for CS-type sulfation can be classified into initial “4-O-sulfation” and “6-O-sulfation” pathways. In the initial step, the non-sulfated O unit [GlcA-GalNAc] serves as a common acceptor substrate for two types of sulfotransferases, chondroitin 4-O-sulfotransferases (C4ST-1 and C4ST-2) 11-13 and chondroitin 6-O-sulfotransferse-1 (C6ST-1), forming monosulfated A (GlcA-GalNAc(4-O-sulfate)) and C (GlcA-GalNAc(6-O-sulfate)) units, respectively (Fig. 2). Subsequent sulfation of A and C units can also occur via GalNAc 4-sulfate 6-O-sulfotransferase (GalNAc4S-6ST) or CS-specific uronyl 2-O-sulfotransferase (UST), resulting in the formation of disulfated disaccharide E (GlcA-GalNAc(4,6-O-disulfate)) and D (GlcA(2-O-sulfate)-GalNAc(6-O-sulfate)) units, respectively (Fig. 2) 9.
Of these CS biosynthetic enzymes, it has been reported that the expressions of ChGn-1, C4ST-1, and GalNAc4S-6ST are upregulated in breast cancer cells 8. Moreover, the gene expression of C4ST-1 has been shown to be positively correlated with progression of breast cancers 14, and CSs produced by C4ST-1 function as a ligand of P-selectin in aggressive breast cancer cells 2. These data suggest that tumor cells drive a change in CS biosynthesis to acquire metastatic capacity.
Recent studies have revealed the possibility that we can fight cancer using oncofetal chondroitin sulfate-binding proteins from malaria parasites 15. Pregnant women are particularly susceptible to malaria infection, because Plasmodium falciparum malaria-infected erythrocytes sequester in the placenta. Scientists have found that A-unit-containing CS (CS-A) expressing in the placenta is the major receptor for adhesion of parasitized red blood cells. In addition, the accumulation of infected erythrocytes in the placenta is caused by a parasite protein of the PfEMP1 (P. falciparum erythrocyte membrane protein 1) family, VAR2CSA (Fig. 3). The VAR2CSA protein is implicated as the adhesion receptor for CS-A expressing only in the placenta, while CSs expressing elsewhere in the body are not recognized by VAR2CSA (Fig. 3). Placental CS-A is a uniquely low-sulfated CS containing only about 2-14% of 4-O-sulfated CS disaccharides (A-units) and 86-98% of non-sulfated CS disaccharides (O-units) 16. Synthesis of VAR2CSA-binding CS-A involves several enzymes. RNAi-mediated knockdown experiments showed that B3GAT1 (GlcAT-I), CSGALNACT1 (ChGn-1), and CHST11 (C4ST-1) are important for synthesis of VAR2CSA-binding CS-A 17. In addition, the 4-O-sulfation level of CS is balanced by 4-O-sulfatase ARSB (arylsulfatase), which removes 4-O-sulfates from the CS chains. Knockdown of ARSB increased the binding of VAR2CSA 17. Therefore, A-units in CS-A chains constitute an important component of the VAR2CSA-binding CS chains. However, no polymorphisms within the genes that code for C4STs influence women’s susceptibility to placental malaria have been reported 18.
The function of low-sulfated CS-A is considered to be associated with the ability of trophoblasts to invade the uterine tissues and promote rapid cell proliferation in the normal placental implantation process 19. Because proliferation and invasion are characteristics of cancer cells, scientists have tried to find hidden connection between placental tissues and cancer. As a result, it has been shown that a similar type of placental CS-A is broadly expressed in malignant human cancer cells 17. Thus, CS-A recognized by VAR2CSA proteins can be used as a pan-cancer cell surface marker. Of note, VAR2CSA proteins show very limited binding to CSs expressing in non-cancerous cells or normal tissues except for placental tissues 17. A previous report has demonstrated that tumor CS-A chains up-regulate tumor-associated integrin signaling through interaction with integrin (Fig. 3) 20. In addition, targeting of CS-A with VAR2CSA could interfere with metastasis because of inhibition of downstream integrin signaling (Fig. 3) 20.
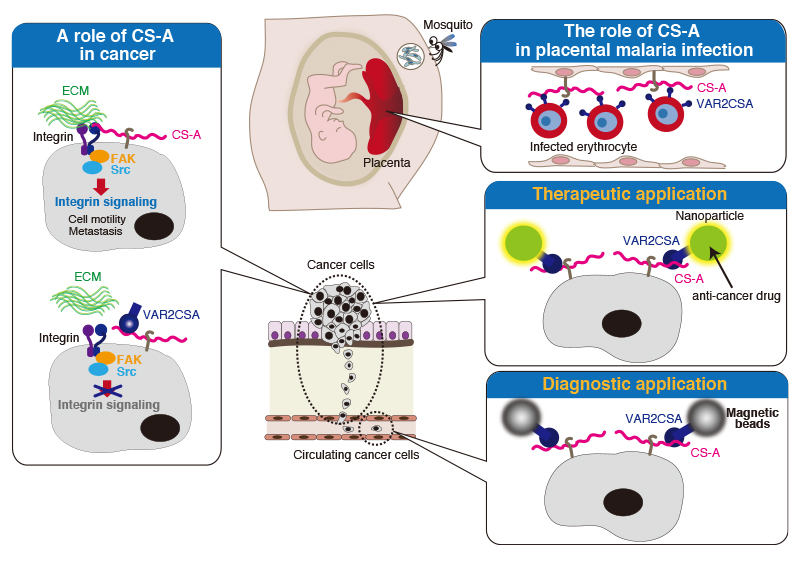
Currently, medical researchers have attempted to eradicate cancer using recombinant VAR2CSA to target cancer-associated CS-A (Fig. 3). In in vivo xenograft animal models, it has been demonstrated that CS-A-binding nanoparticles containing anti-cancer drug target cancer cells using a synthetic CS-A-binding peptide derived from malarial protein VAR2CSA and halt tumor growth 15,21. In addition, novel cancer diagnostics by VAR2CSA approach are under development 22. By capture of circulating cancer cells, taking advantage of the specific interaction between VAR2CSA and tumor CS-A chains, we become able to determine whether a cancer is an aggressive type or not. In the future, a simple blood test will allow the early diagnosis of a wide range of cancers.
Recently, it has been found that CS, which is a dietary supplement approved for use in osteoarthritis, may promote the growth of tumor cells in a type of melanoma 23. Melanoma that starts in melanocytes is the most dangerous type of skin cancer, because melanoma spreads more readily to other parts of the body if not found and treated early. More than 50% of melanomas express the oncogenic BRAF V600E mutation. Although the anti-tumor drug vemurafenib, a BRAF inhibitor, reduces the growth of human melanoma cells that express the BRAF V600E mutation. In the initial stage of treatment, V600E tumors gradually become resistant to vemurafenib, probably because of the activation of an alternative pathway. A recent study has shown that CS-A supplementation mediated BRAF inhibitor resistance in animal experiments using xenograft mice injected with human melanoma cells 23. Therefore, Xia et al. suggested the development of the concept of “precision diet” 24, which should be designed based on individual genetic background to include dietary components and supplements that will lower cancer risk and provide cancer prevention 23.
How does CS-A enhance oncogenic properties of V600E melanoma cells? In the first place, it has been reported that CSGlcA-T, also known as ChSy-3 25, was identified as a “synthetic lethal” partner of BRAF V600E in human melanoma using a systematic RNAi-based screen. This finding suggests that the oncogenic BRAF V600E mutant is vulnerable to synthetic failure of CS. In other words, the CS biosynthesized by CSGlcA-T is needed for V600E melanoma growth. Using a patient-derived xenograft mouse model of melanoma, it has been found that orally administered CS-A resulted in elevated circulation CS-A levels in serum and intracellular CS-A level in tumors 23. Furthermore, it has been shown that increased intracellular CS-A levels elevated the expression of CSGlcA-T 23. CSGlcA-T knockdown resulted in decreased intracellular CS-A levels, leading to attenuated growth rates, masses, and size of tumors. Interestingly, CSGlcAT-I knockdown resulted in decreased cell proliferation in melanoma cells expressing BRAF V600E, but not in cells expressing BRAF wild-type 23. These findings indicate that CSGlcA-T induced by dietary supplement CS-A enhances synthesis of endogenous CS-A in tumors and thus enhances cell proliferation of melanoma cells expressing BRAF V600E. However, how the expression levels of CSGlcA-T are controlled by dietary supplement CS-A that enters cells through endocytosis remains unclear. In addition, further studies are needed to elucidate the molecular mechanism underlying the cancer-type-specific upregulation mechanism of CSGlcA-T in melanoma, because CSGlcAT-I was specifically upregulated by CS-A in melanoma tissues.
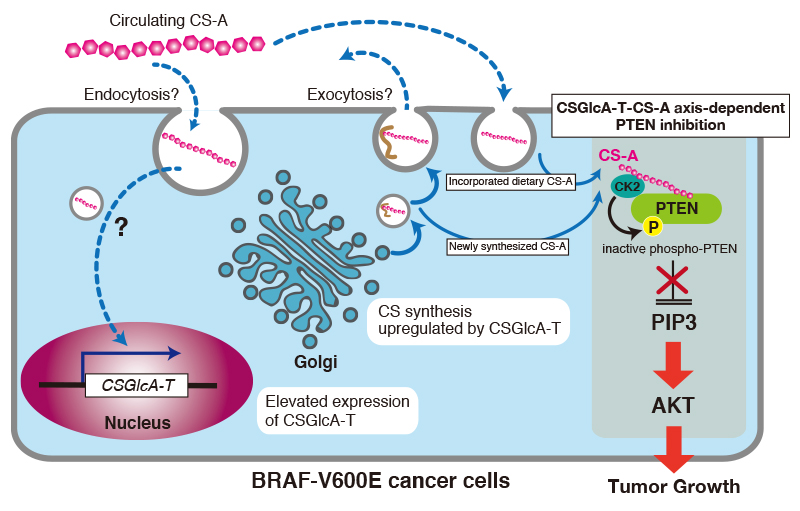
How does CS-A incorporated into tumors and newly synthesized CS-A by CSGlcA-T in tumors exhibit oncogenic activity? Lin et al. have indicated that CS-A maintains PIP3 levels and sustains AKT activation through PTEN inhibition 23. Of note, CS-C is also able to inhibit PTEN in similar way to CS-A. In addition, Lin et al. have reported that CS-A chains function to promote CK2-PTEN interaction and consequent phosphorylation and inhibition of PTEN 23 (Fig. 4). Incidentally, PTEN localizes exclusively to the cytoplasm, where PTEN has no chance to encounter endocytosed CS-A or newly synthesized CS-A. However, recent studies have shown that PTEN can be secreted/exported into the extracellular environment for uptake by recipient cells, within which it functions as an intracellular tumor suppressor 26,27. Thus, PTEN and CS-A might co-localize after endocytosis.
Extensive evidence supports the importance of tumor-associated CSs in promoting aggressive and metastatic behavior of malignant cells 2,28. In addition, CSs present in the ECM of normal tissues also affect tumor cells 7. Of the CS biosynthetic enzymes, it has been reported that the expression of C4ST-1 and GalNAc4S-6ST, which are involved in synthesis of E-units, are upregulated in breast cancer cells 8. Thus, the effects of E-units on breast cancer cells were examined. Interestingly, the invasive activity of the basal-like breast cancer subtypes (MDA-MB-231 and BT-549 cells) was elevated by treatment with chondroitin sulfate E (CS-E) characterized by high E-unit content 29. In contrast, two different types of CS, CS-A and CS-C, did not enhance the invasive activity of BT-549 cells, indicating that CS-E specifically activated the invasion potential of BT-549 cells 29. Furthermore, several analyses were carried out to clarify the molecular mechanism by which CS-E contributes to tumor cell invasion. We previously found that CS-E is a receptor for N-cadherin in osteoblasts 30. In addition, N-cadherin is expressed in highly invasive tumor cell lines, including BT-549 cells, and contributes to the invasive phenotype 14,31. These findings raised the possibility that CS-E transduces signals into breast cancer cells through N-cadherin to up-regulate the tumor invasion potential. As expected, it has been shown that N-cadherin mainly serves as a receptor for CS-E and controls β-catenin signaling via N-cadherin in BT-594 cells 29.
The following cell biological events are involved in transduction of CS-E signals into the nucleus, mediated through N-cadherin: First, N-cadherin is incorporated into the cells by endocytic pathways after interaction of CS-E with N-cadherin. Successively, proteolysis of N-cadherins proceeds in two steps by multiple proteases. This proteolysis process is well known to be a regulated intramembrane proteolysis (Rip), by which transmembrane proteins are cleaved to release cytoplasmic domains that often enter the nucleus to regulate gene transcription 32. In the first step, the extracellular region of N-cadherin is cleaved by multiple proteases, including ADAMs, MMPs, and other transmembrane proteases. Subsequently, a second cleavage occurs within the transmembrane segment by another protease to liberate the cytoplasmic C-terminal fragment (CTF). The N-cadherin CTF can interact with β-catenin. This complex enters the nucleus, leads to β-catenin-dependent transcriptional activation, and can promote cell migration and invasion 33.
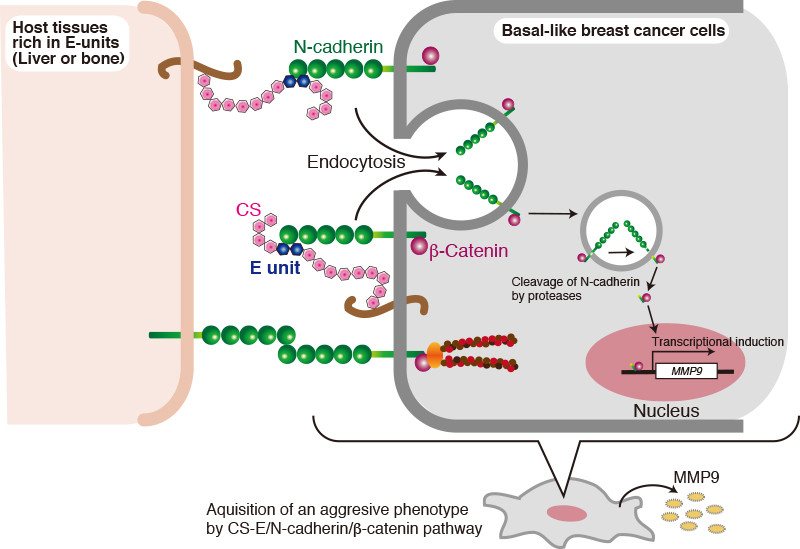
What is the pathophysiological significance of CS-E-induced activation of N-cadherin/β-catenin signaling pathway? Breast cancer mainly metastasizes to the bony skeleton, the lungs, liver, and brain via the circulation. Liver metastasis in particular is a frequent occurrence in patients with breast cancer 34. Liver and bone express CSs and have a high content of CS-E. As shown in Fig. 5, breast cancer cells might bind through their N-cadherin to CS-E in the liver microenvironment to elevate their invasive potential via activation of the N-cadherin/β-catenin pathway. In addition, CS-E that is expressed in cancer cells might form a positive feedback loop, thereby amplifying N-cadherin/β-catenin signaling (Fig. 5). Further studies are needed to elucidate whether cell-autonomous activation of N-cadherin/β-catenin signaling by CS-E contributes to breast cancer malignancy in vivo.
Biological relevance between chondroitin 6-O-sulfates (CS-C) rich in C-units and inflammation are speculated, because CS-C could influence the recovery process after nerve injury and the pathological condition of neuroimmunological diseases. For example, the expression of C6ST-1 and its products, CS-C, are upregulated after central and peripheral nerve injury, and CS-C exhibits a positive influence on axonal regeneration 35. In addition, phenotypes of mice subjected to experimental autoimmune encephalomyelitis are affected by the expression levels of C6ST-1 36. It is commonly accepted that cancer development and progression are tightly linked to a chronic low-grade inflammation, which is characterized by tissue infiltration by innate and adaptive immune cells. The local environment, or niche, of cancer cells plays multiple roles in tumor inflammation. It has been reported that C-units upregulate in human cancer stroma, while A-units decrease compared to healthy tissue 37. In cancer cells, the expression of C4ST-1 is upregulated and the expression of A-units is higher compared with normal cells 8,38. Soluble CS-C functions as a scavenger of nitric oxide, a pro-inflammatory mediator released from macrophages reside in the tumor niche 39. In addition, CS-C significantly reduces production of pro-inflammatory cytokines, interleukin-6, and tumor necrosis factor-α, in interferon-γ/lipopolysaccharide (LPS)- or LPS-activated macrophages 39. These anti-inflammatory effects of CS-C are responsible at least in part for suppression of NF-κB nuclear translocation 39,40. These findings suggest that CS-C inhibits the broadest spectrum of inflammatory mediators, and thus, CS-C accumulated in the tumor niche can affect the profile of cytokines secreted from tumor niche-resident macrophages. Because typical proinflammatory cytokines produced by the type M1 macrophage manifest protumor activity at the initial stages of the development of tumor, CS-C might serve as an anti-tumor regulator. Further studies are needed to clarify the role of CS-C in cancer biology.
The accumulated knowledge regarding the altered structure of CSs in cancer indicates their importance as biomarkers for disease diagnosis and progression. In addition, CSs themselves transmit signals or function as co-receptors of growth factors. In recent years, cell biology studies have revealed the action mechanisms of CSs in cancer biology as described above.
The most recent study has shown that CA19-9, a conventional glycan marker for pancreatic cancer, directly led to pancreatitis and accelerated pancreatic cancer in mice 41. Elevated levels of CA19-9 recruits the immune cells to induce a cascade of inflammation reactions propelled by the release of deleterious digestive enzymes from the pancreas, leading to severe pancreatitis 41. By blockage of the function of CA19-9 with antibodies, pancreatic damage could be prevented in mice models 41. From this finding, CA19-9 is a promising therapeutic target for pancreatitis. As is the case with CA19-9, if we can find CS structures for which changes are directly linked to functional consequences, we will be able to develop new medicines.



