
Kota Nomura
Ph.D. student, Laboratory for Organic Biochemistry (Prof. Yasuhiro Kajihara), Department of Chemistry, School of Science, Osaka University.
Kusumoto Award 2018, 101st The Chemical Society of Japan Student Presentation Award 2021, 21st Kansai Glyco Science Forum Best Presentation Award 2021, 40th Japan Society of Carbohydrate Research Excellent Presentation Award 2021, and 12th Otsu Conference Award Fellow, MSD Life Science Foundation 2021.
To understand the biological processes mediated by various glycoforms of glycoproteins, the preparation of homogeneous glycoproteins is essential for performing extensive biological experiments. Recently we found that diacyl disulfide coupling (DDC) between glycosyl asparagine and peptide yielded glycopeptides chemoselectively. DDC enabled us to develop a new glycoprotein synthesis method, “the chemical glycan insertion strategy,” which can insert glycosyl asparagine between the N- and C- termini of two unprotected peptides. In our design concept, fewer steps are needed to generate glycoproteins. Herein, I will introduce the latest strategy of glycoproteins synthesis by chemical insertion.
Post-translational modifications (PTMs) of proteins are essential for protein activation. According to recent research, PTMs regulate a wide range of biological processes, including several disease processes such as cancer, Alzheimer’s disease, and Parkinson’s disease1-4. Of these protein modifications, glycosylation is one of the most common modifications. Glycoproteins are found on the cell surface and in body fluids, and these proteins are modified with serine/threonine-linked O-glycans or asparagine-linked N-glycans5 (Figure 1A, B). The biosynthesis of glycans is regulated by the substrate specificity of glycosyltransferases and glycosidases, resulting in considerable diversity of glycan structures (glycoforms)6 (Figure 1C). Under these circumstances, identifying which glycans play an important role in sustaining specific biological events is impossible7-10.
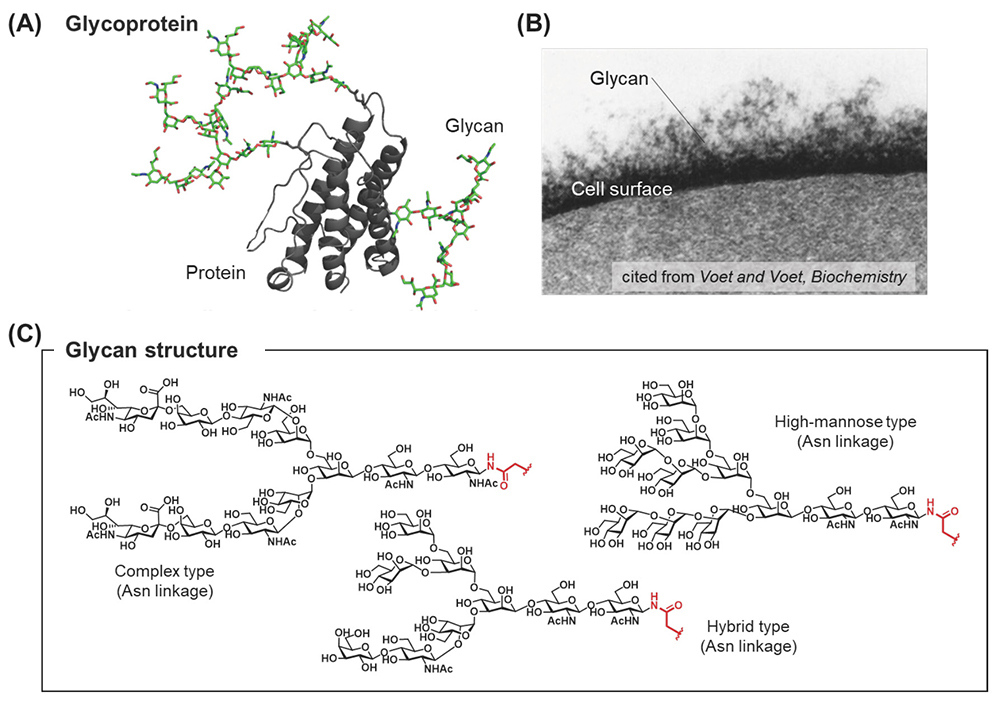
To understand the biological processes mediated by each glycoform, the preparation of homogeneous glycoproteins is essential for carrying out extensive biological experiments6. One of the most popular glycoprotein preparation methods is total chemical synthesis using native chemical ligation (NCL)11. Chemical methods involve the preparation of both glycopeptides 2 and peptides 3 by solid phase peptide synthesis (SPPS)12 using glycosyl amino acid derivatives 1 (Figure 2A), and those segments are sequentially coupled by NCL to afford full segments of glycoproteins 5 (Figure 2B).
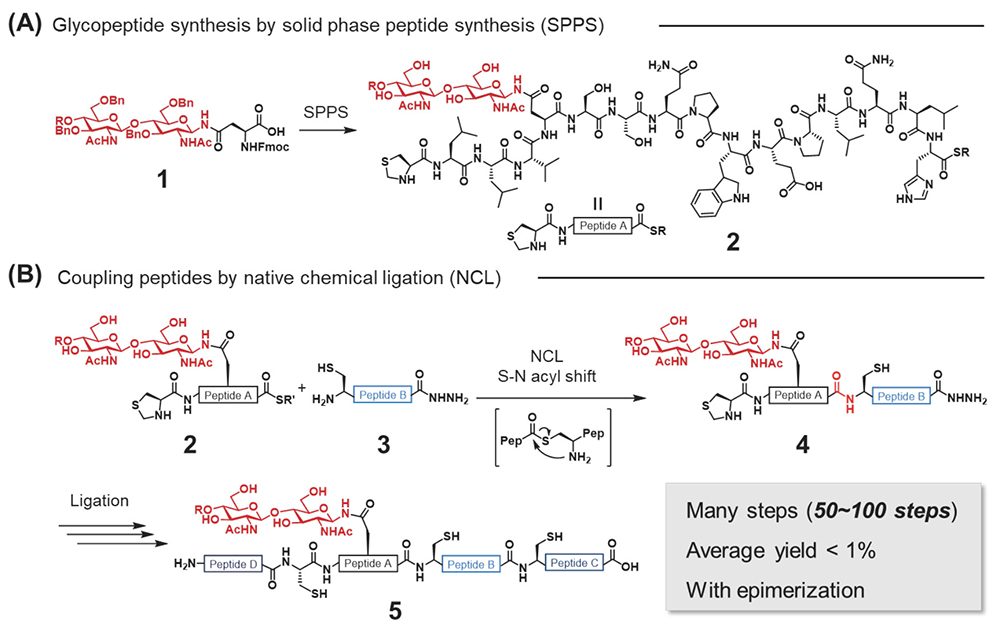
However, conventional chemical approaches are relatively time-consuming, requiring multiple synthetic steps and an appropriate amount of valuable N- or O-glycans because glycopeptide syntheses are set at the early stage of total synthesis (Figure 1). To accelerate the elucidation studies of glycan functions, we need to solve these synthesis problems.
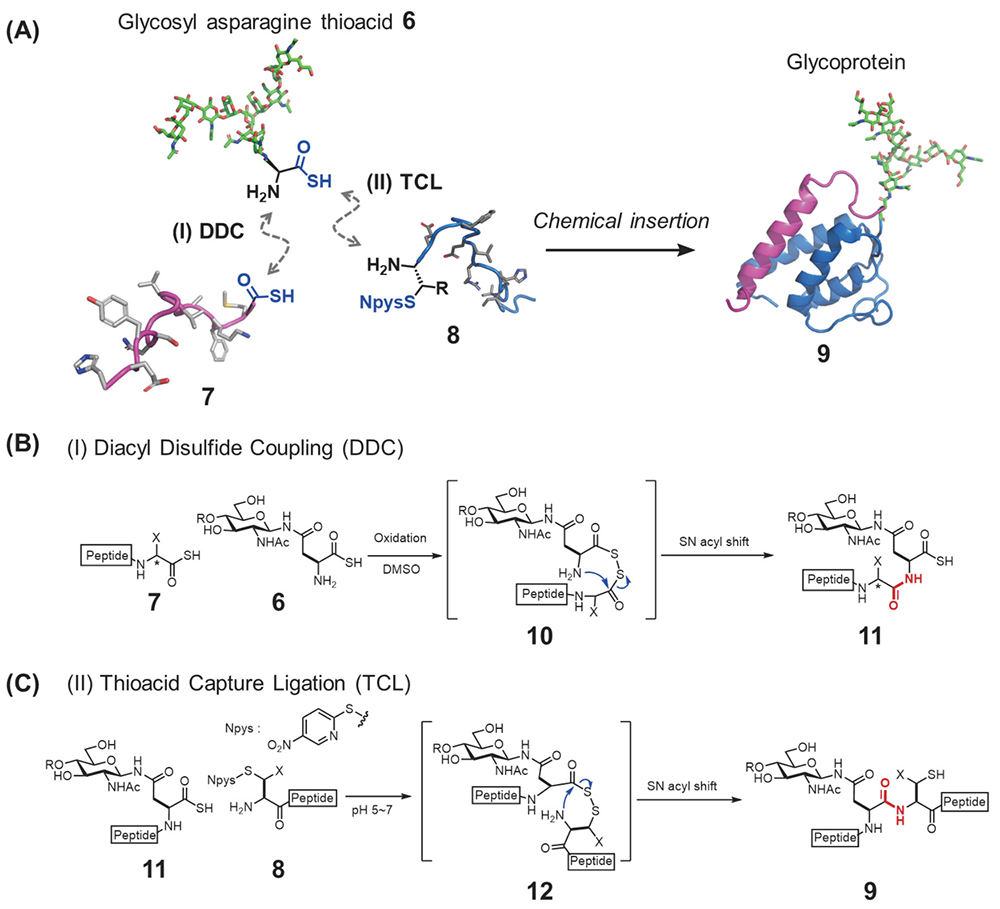
We developed an alternative synthesis method, a highly convergent glycoprotein synthesis strategy designated the “chemical glycan insertion strategy,” that entails the coupling of two peptides 7,8 with the N- and C-termini of glycosyl asparagine thioacid 6 chemoselectively13 (Figure 3A). Thioacids were originally used as acylation functionalities for condensation reactions14 and our group also developed chemoselective amide formation reactions based on thioacid15,16. In the chemical glycan insertion strategy, the first coupling is diacyl disulfide coupling (DDC) between a peptide thioacid 7 and glycosyl asparagine thioacid 6 (Figure 3B)13. Because the resultant glycopeptide 11 has a thioacid form at its C terminus, we could apply thioacid capture ligation (TCL)17,18 for the coupling of the resultant glycopeptide thioacid 11 and another peptide 8 having a nitropyridyl disulfide functional group (Npys) at its N-terminus to afford the full-length glycoprotein backbone 9 (Figure 3C). This route enables us to assemble the entire glycoprotein backbone in just two steps. In addition to this efficiency gain, valuable glycosyl amino thioacids 6 can be used at the late stage of the synthesis of glycoproteins.
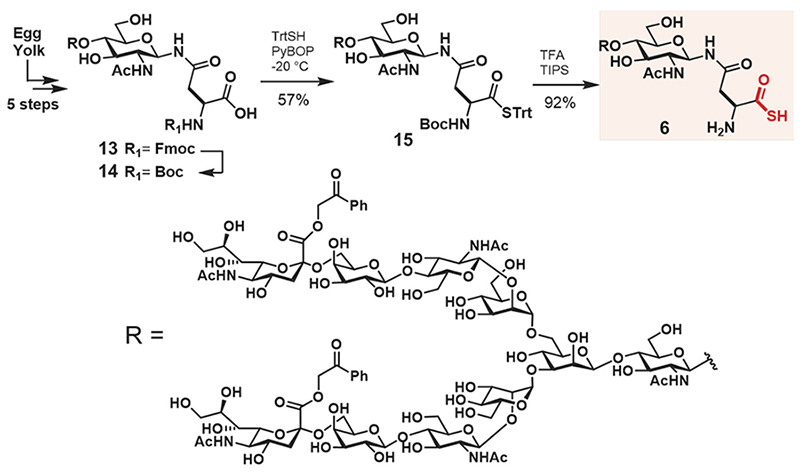
The synthesis of glycosyl asparagine thioacid 6 is shown in Scheme 1. The Boc protected substrate, glycosyl asparagine 14, was prepared by the reported method19, and the condensation of trityl thiol was performed with PyBOP at −20°C to give compound 15 (57% yield). Finally, deprotection of both the Boc group and trityl group was performed with trifluoroacetic acid, and triisopropylsilane yielded the desired glycosyl asparagine thioacid 6 (92% yield). The structure was confirmed by nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS).
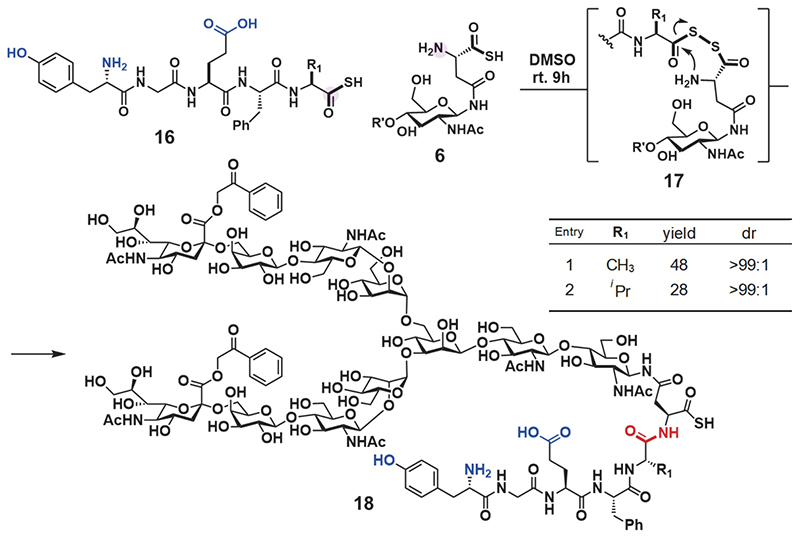
Next, we employed glycosyl asparagine thioacid 6 and peptide thioacid 16 for DDC. The product of two equivalents of glycosyl asparagine thioacid 6 toward peptide thioacid 16 (one equivalent) reacted with various amino acids at the C-terminus in dimethyl sulfoxide (DMSO) at room temperature. The reaction velocity was slow, but the desired glycopeptide thioacid 18 formed as a major product after 9 h (Scheme 2). The reaction proceeded in about 30% yield even when a bulky amino acid such as valine was present at the end of the peptide 16. Furthermore, liquid chromatography (LC) and nuclear magnetic resonance (NMR) analysis confirmed that epimerization does not proceed in the condensation reaction (Scheme 2).
We have so far achieved the synthesis of several biochemically important cytokines by using the newly developed glycan insertion strategy. In this article, I introduce the following two synthetic examples reported in our recent publications1 (Figure4, 5).
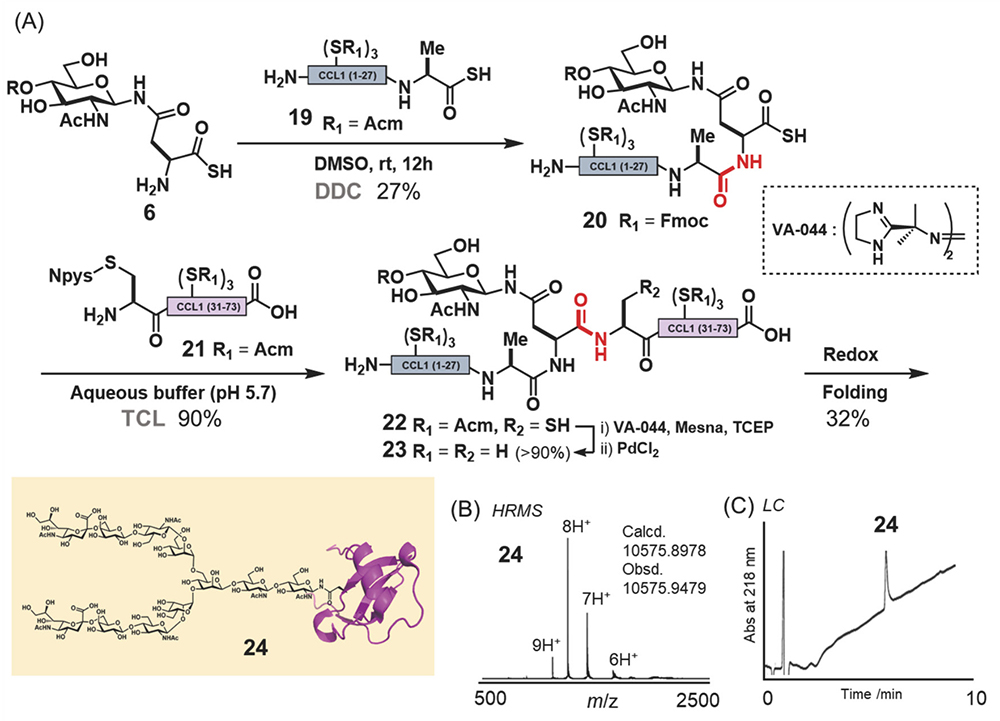
First, we synthesized CCL1 24 with a sialyloligosaccharide. CCL1 has a complex-type, N-linked sialyloligosaccharide at the 29th position20,21. According to the chemical insertion strategy (Figure 3A), CCL1 24 was separated into three components: glycosyl asparagine thioacid 6, peptide thioacid 19, and peptide derivative having an Npys group 21 (Figure 4A). Glycopeptide thioacid 20 was obtained by the DDC of 2 equivalents of peptide thioacid 19 and glycosyl asparagine thioacid 6 (15 mM), with a 27% isolated yield in DMSO. After isolation of glycopeptide thioacid 20, glycopeptide 20 and 2 equivalents of peptide derivative 21 (1.0 mM) were coupled by TCL in a buffer solution (0.2 M sodium phosphate, pH 5.7) containing 6 M guanidine-HCl to yield the protected full length CCL1 peptide 22 (>90% isolated yield). Desulfurization of the 30th cysteine with a radical initiator and subsequent deprotection of the Acm protecting groups of cysteines with PdCl2 and the phenacyl protecting group of sialyloligosaccharide with piperidine and 2-mercaptoethanol (BME) were performed to yield glycosyl CCL1 polypeptide 23. Finally, oxidative folding under redox conditions at pH 8.0 yielded CCL1 24 with a sialyloligosaccharide at the 29th position. After isolation of the folded CCL1 24, the LC spectrum (Figure 4C) and HRMS (Figure 4B) supported the correctly folded structure of glycosyl CCL1 24.
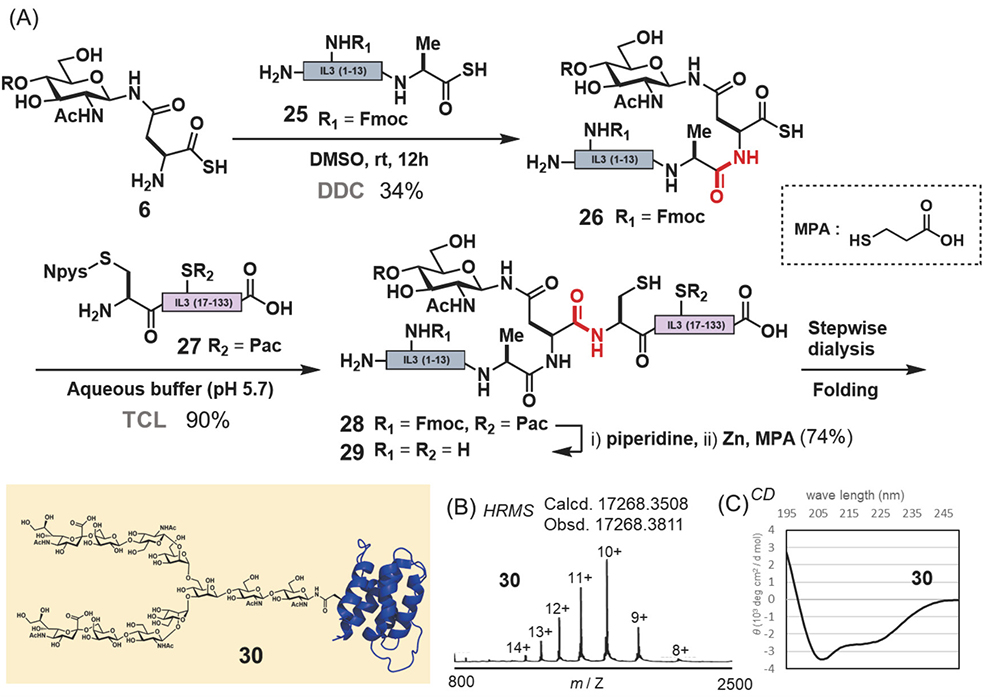
Next, we set out to synthesize IL3, a cytokine produced by T cells as a regulator of hematopoiesis22−24. Glycosylated IL3 is an ideal target to apply our strategy, as there are no synthetic examples to date. For the synthesis of IL3 using the chemical insertion strategy (Figure 3A), we employed glycosyl asparagine thioacid 6 and two peptides: peptide thioacid 25 and peptide derivative having an Npys group 27 (Figure 5A). Furthermore peptide 27 was prepared in >40% yield (total 4 steps) by an E.coli. expression system and several modification steps, including pyridyl disulfide formation of the N-terminal thiol. The DDC of 2 equivalents of peptide thioacid 25 with glycosyl asparagine thioacid 6 yielded glycopeptide thioacid 26 (34% isolated yield). Then, the second ligation was performed with 2 equivalents of 26 and peptide 27 (1.0 mM) by TCL in a buffer solution (0.2 M sodium phosphate, pH 5.7) containing 6 M guanidine-HCl to obtain full length IL3 peptide 28 (>90% isolated yield). The formyl protecting group and Fmoc protecting group were removed from the full length glycosyl IL3 28 with piperidine, and then the internal phenacyl group was removed by zinc reduction affording glycosyl IL3 polypeptide 29. The oxidative folding of 29 afforded folded glycosyl IL3 30. After isolation of the folded glycosyl IL3, CD spectra (Figure 5C) and HRMS (Figure 5B) were obtained. The in vitro bioassay of glycosyl IL3 30 was based on the proliferation of TF-1 cells and the activity of 30 was confirmed to be like that of commercially available non-glycosylated IL3 expressed in E.coli. The biological assay and analytical data, such as the CD spectrum and HRMS, supported that the synthetic glycosyl IL3 30 employed had the native folded structure.
We developed a novel and efficient method for synthesizing glycoproteins. The chemical glycan insertion strategy based on DDC allows for the synthesis of the full-length glycoprotein within only 2 steps. Because peptides can be prepared by the E.coli expression system, we will be synthesizing homogeneous glycoproteins more easily soon. Currently, we are further investigating the convergent synthesis of cytokines and cancer-related glycoproteins with complex structures by using our glycan insertion strategy.
The authors thank all of the collaborators who were involved in the research. All work described here was supported by grants from the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Numbers JP20J20649, 21H04708) to K.N. and Y.K., respectively, and from AMED (Grant Number 19ae0101033h0004) to Y.K. The authors gratefully acknowledge this financial support.



