
Hiromune Ando
Professor at the Institute for Glyco-core Research, Gifu University
In 1999, completed his doctorate at the United Graduate School of Agricultural Science, Gifu University; in the same year, became Japan Science and Technology Corporation CREST postdoctoral researcher; in 2002, awarded the Japan Society for Promotion of Science Research Fellowship for Young Scientists; in 2003, appointed Assistant Professor at the Life Science Research Center, Gifu University; in 2008, promoted to Associate Professor in the Faculty of Applied Biological Sciences, Gifu University and the Institute for Integrated Cell-Material Sciences, Kyoto University; in 2017, named Professor at the Center for Highly Advanced Integration of Nano and Life Sciences, Gifu University Institute for Advanced Study and Visiting Professor at the Institute for Integrated Cell-Material Sciences, Kyoto University Institute for Advanced Study. Has held his present post since 2020.
He is studying the creation and application of molecules in order to further elucidate glycan functions involved in the chemical synthesis of glycolipids containing sialic acid.
Sialic acid is a “uronic acid”, sugar with a carboxyl group as an acidic functional group. While many monosaccharides in glycans in the body are pentoses and hexoses, sialic acid is a nonose and plays various and important physiological roles. In the author’s opinion, sialic acid must be a distinguished from other sugars. Regarding the chemical synthesis of glycans, the presence of sialic acid in a glycan dramatically increases the difficulty of its synthesis. Because of its unique structure, sialic acid markedly decreases the success rate of glycosidation. A fundamental solution to this problem has been sought for more than half a century. Recently, our group successfully developed one approach to solving this problem. In this article, I will outline this approach.
In many chemical glycosidation methods, glycosidation is achieved artificially by making a carbon at the reducing terminal of a sugar (anomeric carbon) electron-deficient and then letting an oxygen of a sugar hydroxyl group, which is electron-rich, attack the carbon, thereby producing a new C-O bond. In this type of reaction, successful bond formation depends on the chemical characteristics of the former called the glycosyl donor and the latter called the glycosyl acceptor. Regarding the glycosidation of sialic acid, the oxocarbenium ion of sialic acid, which is an electron-deficient species, has characteristics that are problematic (Figure 1). Although the anomeric carbons to which hydrogens are bound (present for example in glucose and galactose) are not sterically restricted, that of sialic acid is sterically hindered by the carboxyl group. This makes the attack of hydroxyl group difficult. Moreover, the carboxyl group, which is electron‐withdrawing, destabilizes the oxocarbenium ion. More inconveniently, hydrogens of a methylene carbon adjacent to the anomeric carbon tend to be eliminated as cations to compensate the electron deficiency, resulting in a competing inactivation reaction that degrades the oxocarbenium ion to a molecule with double bond (2,3-ene). These three problems are fundamental stumbling blocks to the glycosidation of sialic acid. Even if hydroxyl groups can attack the anomeric carbon, two stereoisomers may be generated. Without directing stereoisomer generation, there is a strong tendency to preferentially generate a β-glycoside due to the anomeric effect, which has not been found in sugar chains in the body. Therefore, some ingenuity is needed to obtain an α-glycoside, the other natural form. However, it is extremely difficult to utilize participation by neighboring nucleophilic substituents in sialic acid, because sialic acid has no hydroxyl group at neighboring positions of the anomeric carbon. In terms of the difficulty in the glycosidation of sialic acid, this is a final blow. The challenge with the α-glycosidation of sialic acid is, so to speak, overcoming this quadruple whammy. Saying in a little exaggerated manner, if glycosidation by Fischer is the beginning of chemical glycosidation, the α-glycosidation of sialic acid is considered to be a challenge in the field of sugar chemistry in approximately 120 years.
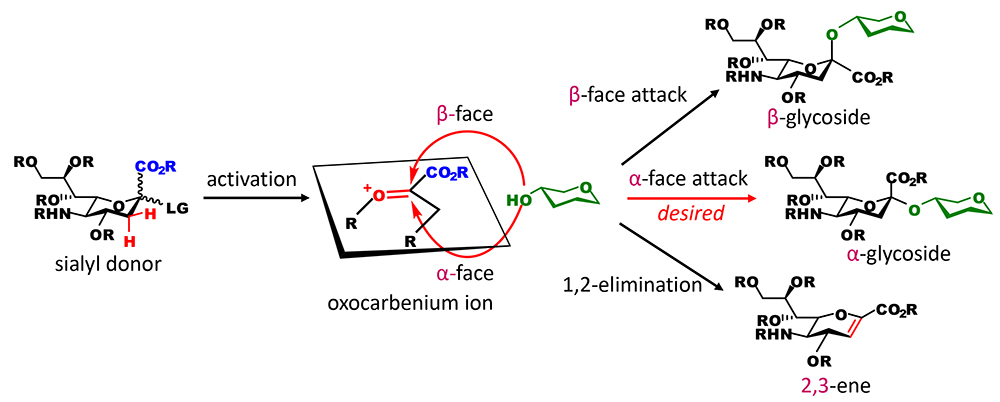
In order to achieve successful α-glycosidation of sialic acid, it is important to suppress the degradation of an unstable oxocarbenium ion intermediate of sialic acid and restrict the attack by hydroxyl groups to the α-face. For effective and α-selective sialidation (glycosidation of sialic acid), various approaches have been developed such as a method using the nitrile solvent effect, a method that introduces auxiliary groups at neighboring positions of the anomeric carbon, and a method that introduces a cyclic carbamate structure at the C4 and C5 positions (for details, please refer to our article)1. The approaches described above were first developed by researchers in Japan. All of these methods stabilize the oxocarbenium ion by blocking the β-face of the oxocarbenium ion with weak binding ligands in order to obtain an α-glycoside, and they have been used to synthesize many glycans containing sialic acid. However, the β form can be generated in principle, and stereoselectivity varies depending on reaction conditions. In terms of practicality, the generation of stereoisomers requires the separation of sialoside anomers with a very similar polarity. This problem should be overcome completely. Therefore, I think the development of a sialidation method that can be used to generate a product with no isomers in principle, that is, a completely α-selective sialidation approach, is very significant.
A carboxyl group at the C1 position, which is a characteristic of sialic acid and causes interference with glycosidation, and an amino group at the C5 position are in 1,4-cis configuration when the stereochemistry of anomeric carbon is α. We anticipated that if these groups could be tethered, the β-face would be completely blocked with the cross-linked part when glycosidation occurs, resulting in the generation of an α-glycoside exclusively (Figure 2).
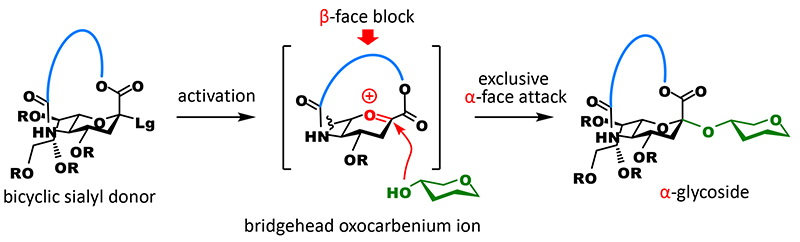
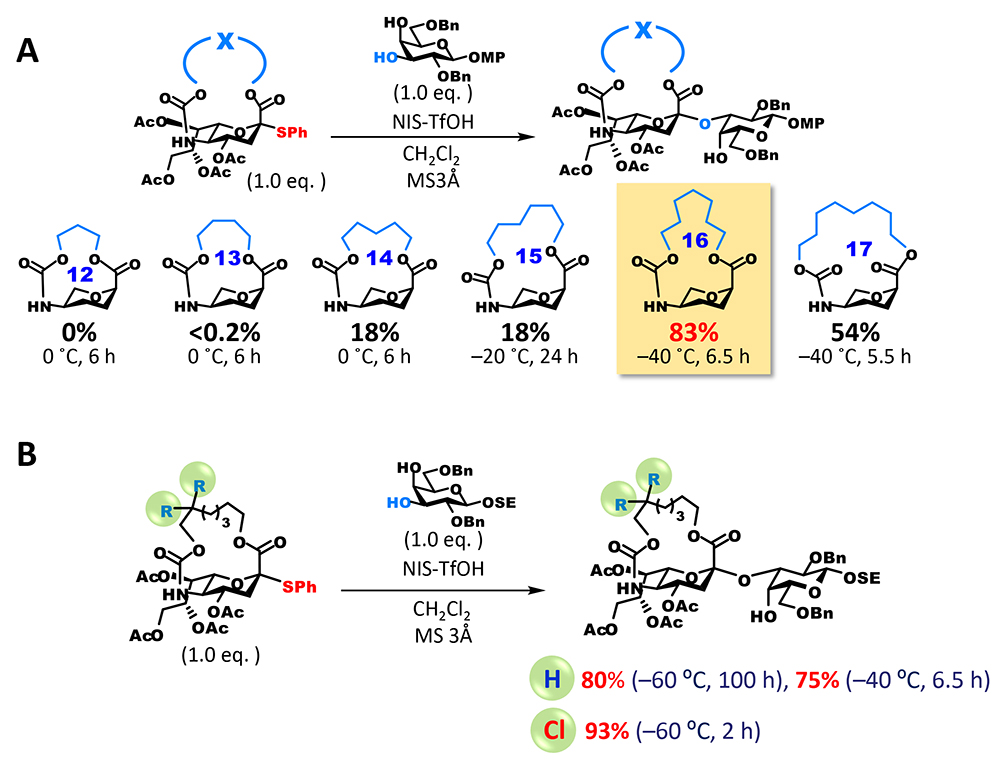
However, the anomeric carbon becomes a bridgehead of the fused ring (one of two binding points of two rings) and strongly distorts the ring, which is overwhelmingly unfavorable for the generation of carbocations that exist as planar sp2 carbons (Bredt’s rule). On the other hand, there are many reports of the generation of anti-Bredt cations or olefins that have macrocyclic structure3,4. We thought that these suggest sp2 carbons could be generated at bridgehead positions by adjusting the length of the tether part. However, the tethered form of sialic acid becomes a bicyclic compound containing aza and oxacyclic groups, and the structure of anti-Bredt cations (or olefins) at bridgehead positions of a fused heterocycle had never been known in literature. Therefore, we decided to experimentally check the length of the tether part that allows the formation of sp2 carbons (Figure 3A). We synthesized candidate compounds for sialyl donor that had a 12 to 17-membered macrocycle by introducing the tether part as a carbonate ester into an amino group at the C5 position and then cyclizing through dehydration condensation between a carboxyl group at the C1 position and a hydroxyl group at the end of the tether part. Subsequently, we investigated glycosidation. As expected, all of the generated glycoside products were α-glycosides and the highest yield was achieved with a donor that had a 16-membered ring. Interestingly, one difference in the number of members of rings greatly affected yield: with a 15-membered ring, glycosidation did not terminate and 39% of the recovered products were unreacted donors, while with a 17-membered ring, glycosidation terminated but approximately 40% of the obtained products were olefins generated through 1,2-elimination (2,3-ene). It can be presumed that glycosidation generating sp3 carbons is preferred because the 16-membered ring is less distorted than the 15-membered ring and allows the generation of cations at bridgehead positions, and the completely planar configuration of the anomeric carbon of the 16-membered ring is energetically unfavorable compared with the configuration of the 17-membered ring.
Although the yield of glycosidation with the 16-membered ring was satisfactory, there was room for improvement because reaction time was relatively long. A carbamate group at the C5 position can be removed by alkaline hydrolysis or other methods. However, taking the synthesis of naturally occurring sialic acid analogs into account, we thought that an improvement enabling the selective release of amino groups was needed. To fulfill these two requirements, we focused on a 2,2,2-trichloroethoxycarbonyl (Troc) group, which is used to protect amino groups. Troc groups can be selectively removed in the presence of other protecting ester groups. We also found that the introduction of a Troc group into the amino group at the C5 position of sialic acid dramatically improves its reactivity as a donor5. To add this characteristic to the macrocyclic structure, a modified form in which the β position of an alkyl chain binding to the carbamate site is dichlorinated was synthesized. As expected, the reactivity of the new dichlorinated donor was much higher than that of the unsubstituted donor, the reaction completed quickly and under mild conditions, and the yield of glycoside was extremely high (Figure 3B). In addition, selective conversion of the carbamate site to a free amino group is possible and can be applied to the synthesis of various sialic acid analogs that have different chemical modifications of the amino group at the C5 position.
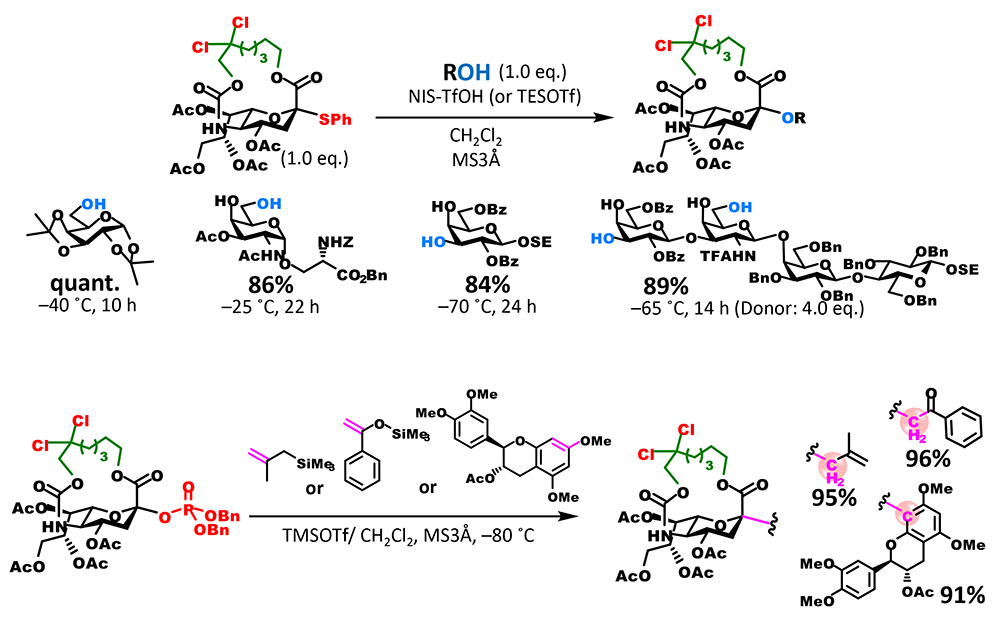
Experiments investigating the substrate scope to which donors are applicable showed that chlorinated donors had a broad substrate scope (Figure 4). By substituting the leaving group with a dibenzyl phosphate ester group, substrates containing olefins, which are damaged under conditions of thioglycoside donor activation, can also be used. Moreover, reaction between a thioglycoside donor and a carbon nucleophile enabled successful generation of a C-glycoside, further expanding the range of substrates to which donors are applicable.
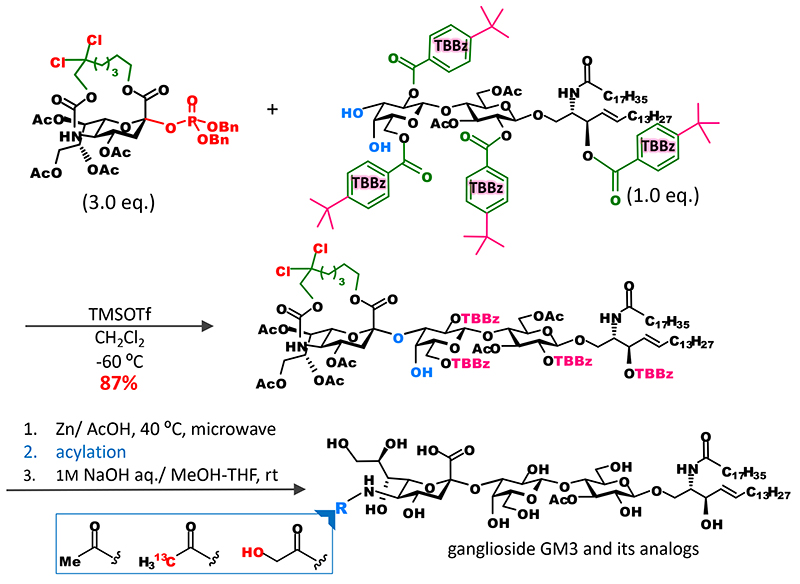
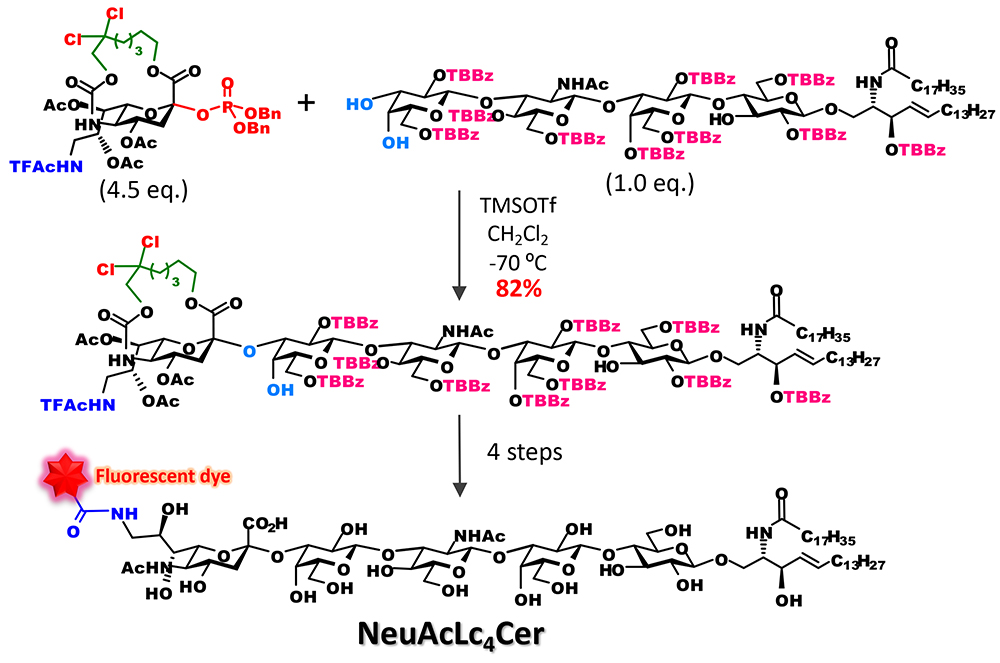
Noteworthily, it became possible to efficiently bind sialic acid to oligosaccharide acceptors. Conventional synthetic methods are used to bind a sialic acid to a monosaccharide and incorporate them as a disaccharide block into a glycan to avoid the separation of stereoisomers. With the new approach, however, it became possible that the introduction of sialic acid that would bind to the end of a sugar chain is performed at the final stage of chain elongation. For example, we were able to show that various analogs of ganglioside GM3 can be synthesized as follows: directly by glycosidation of sialic acid and introduction of it into glycolipid acceptor to obtain the skeleton of ganglioside GM3; and then selective cleavage of the tether part and acylation of an amino group at position 56 (Figure 5). Moreover, we applied this approach to the synthesis of fluorescent probes of ganglioside with a long sugar chain and were able to develop a lacto-series ganglioside fluorescent probes for single-molecule imaging7 (Figure 6).
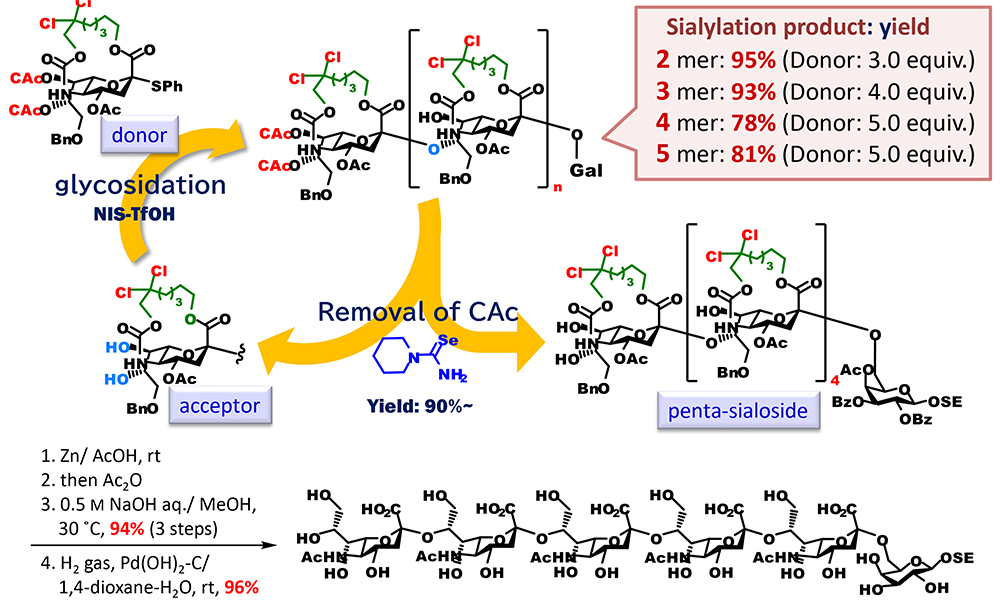
Moreover, we were able to show the usefulness of an approach using tethered sialic acids for the synthesis of sialic acid polymer, which is difficult to synthesize (Figure 7). Naturally occurring glycans contain the tandem structure of sialic acids. From the viewpoint of synthetic chemistry of glycans, the most difficult structure to synthesize is an α(2,8)-linked oligosialic acid, which is sialic acids crosslinked through hydroxyl groups at the C8 position. It is presumed that this is caused by markedly decreased reactivity of the hydroxyl group at the C8 position of sialic acid due to hydrogen-bonding with the amido group at the C5 position. We anticipated that macrocyclization increases a barrier to free rotation of the bond between a carbon at the C4 position and a nitrogen at the C5 position, which is unfavorable for ring formation by hydrogen-bonding between the hydroxy group at the C8 position and the NH group at the C5 position. On the basis of this, we synthesized an acceptor that had the same ring-fused structure as that of the donor and had free hydroxyl groups at the C7 and C8 positions and used it for glycosidation with the donor. As expected, we successfully synthesized a sialic acid dimer with α(2,8)-linkage in good yield. The conformational analysis of acceptors using density-functional theory (DFT) calculation revealed that the acceptors mainly have not only the usual chair conformation, which is 2C5, but also its inverted version, the 5C2 chair conformation, and the 2,5B boat conformation. On the basis of this result, we attempted to synthesize α(2,8)-linked oligosialic acid by synthesizing a donor, protecting the hydroxyl groups at the C7 and C8 position with a selectively removable chloroacetyl groups, and then repeating two processes, which were glycosidation and conversion to an acceptor by selective removal of the chloroacetyl groups. As a result, we successfully elongated it to a pentamer, and the obtained pentamer was converted to an unprotected form through the selective cleavage of the tether part, N-acetylation, saponification, and the removal of the benzyl groups.
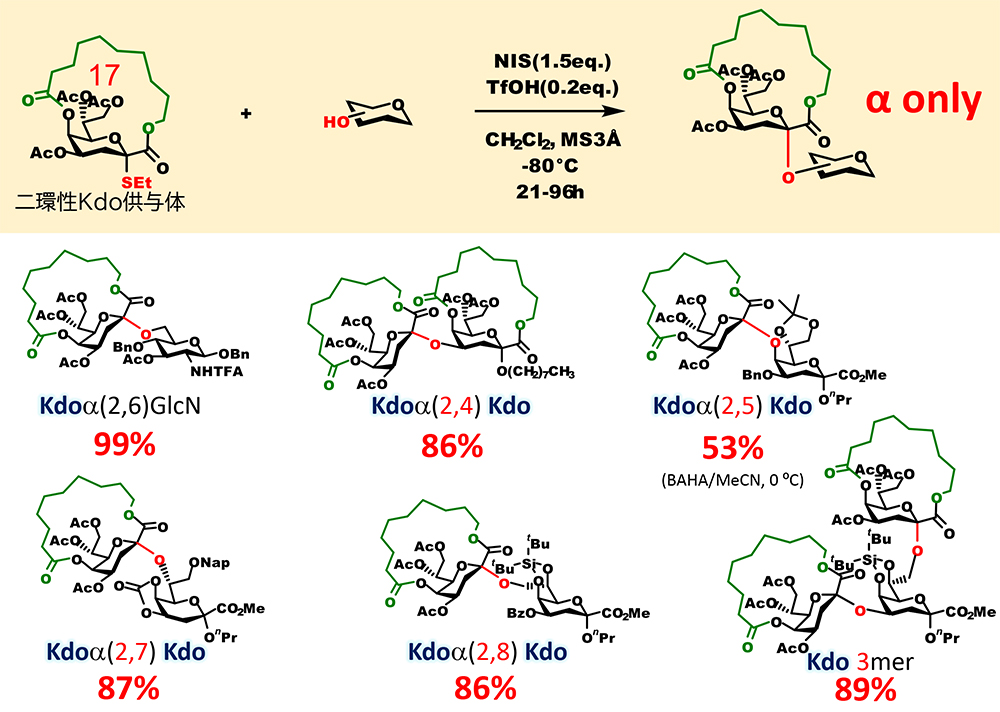
Recently, we were also able to apply the above-mentioned stereocontrolled glycosidation using bicyclic macrocyclic structure to the α-glycosidation of 2-keto-3-deoxyoctulosonic acid (Kdo), which has a structure similar to sialic acid8,9 (Figure 8). Kdo is a sugar residue constituting lipopolysaccharides and capsular polysaccharides, which are located outside the membrane of bacteria, and glycans containing Kdo are involved in bacterial infection and immune evasion within the host. Highly reliable methods for permanently obtaining stereoselectivity in α-glycosidation of Kdo have been developed. However, there has been no mechanism to completely eliminate the generation of a β-isomer, like the one for sialic acid. Our investigation of the structure of macrocyclized Kdo found that a donor with a 17-membered macrocyclic structure and an ethylsulfonyl leaving group is the most suitable. Using this bicyclic Kdo donor, we succeeded in stereoselectively synthesizing Kdo tandem structure seen in sugar chains of bacteria.
As described above, using a bridgehead oxocarbenium ion as an electrophilic species, we realized completely stereoselective α-glycosidation of sialic acid, which was previously impossible. It can be said that the glycosidation of sialic acid, which was previously considered the most difficult, now became the easiest glycosidation (in the sense that no stereoisomer is generated). This is expected to streamline sialo-glycan synthesis and the conjugation of sialic acid and other functional molecules. Since the problem of stereocontrol in the glycosidation of sialic acid is also a bottleneck in the automation of sugar chain synthesis, it is expected that application of this reaction will spur further development. In a similar manner, we also realized completely selective α-glycosidation of Kdo using a bridgehead oxocarbenium ion as a reactive species. G;ycan synthesis is indispensable to propel the elucidation of biological functions of sugar chains and application of these functions to the development of medicines and other substances. Thus, it is important to continue to advance and strengthen the technical basis. The glycosidation methods described in this article need further improvement. We would like to apply these methods to development of sufficiently practical approaches.



